Lithium Inventory is a new initiative, started in 2023, intended as a free and open knowledge hub for battery science and electrochemistry, written by practitioners for practitioners.
Info
This venture is in a very early stage and the majority of the content will come later. In the meantime, you can read the principles on which this site is founded and intended to become, as well as the Content Roadmap.
If you are interested in contributing, please reach out to Matt.
Lithium Inventory now comprises 20712 words across 23 pages.
Subsections of Lithium Inventory
Chapter 1
Battery fundamentals
In Battery Fundamentals we will introduce the chemistry and electrochemistry of batteries, first in a general sense, and with a specific focus on lithium-ion (Li-ion) batteries.
In Introduction to Battery Chemistry we will look at the fundamentals operating principles of batteries in a general sense, introducing some of the core concepts in electrochemistry.
On this page we’ll look at the origin of cell voltage using a classic battery system, the Daniell cell, as an example. We’ll look at the concepts of redox couples, standard electrode potentials, and the Nernst equation, and predict the discharge voltage curve for the Daniell cell.
Half reactions
Let’s start with a very simple example of a battery: the Daniell cell. This battery uses a negative electrode of zinc metal, immersed in a solution of a zinc salt, and a positive electrode of copper metal, immersed in a solution of a copper salt. Between the electrodes is a porous separator, which also separates the two salt solutions, but allows the transfer of ions between the two electrodes. A schematic of the Daniell cell is shown below.
If there is no external electronic connection between the electrodes, there is an equilibrium between the metal and the ions in the electrolyte, which we can write as a half reaction, in which the metal is the reduced form, and the dissolved ion of that metal is the oxidised form. Any pair of corresponding reduced and oxidised species is referred to as a redox couple. For each redox couple, there is an associated electrode potential, which describes the relative power of the reduced form to act as a reducing agent; that is, its tendency to release its electrons to become its oxidised form. For the Daniell cell, we have the following half-reactions:
At the negative electrode:
$\ce{Zn^{2+} <=> Zn + 2e-}$, with a standard potential of -0.34 V vs SHE.
At the positive electrode:
$\ce{Cu^{2+} <=> Cu + 2e-}$, with a standard potential of +0.76 V vs SHE.
The word “standard” is very important here. “Standard” means “measured under standard conditions”, which is defined as 1 mol dm-3 of dissolved species in aqueous solution, 1 atm of pressure, 25 °C. SHE means “standard hydrogen electrode”, which is the universally accepted “zero” against which standard potentials are measured. In practice, we’re usually not dealing with standard conditions, or standard electrodes, and we will deal with the consequences of this later.
The lower standard potential of the Zn/Zn2+ redox couple compared to the Cu/Cu2+ redox couple tells us that Zn metal is a stronger reducing agent than Cu; in turn, this tells us that Zn will spontaneously reduce Cu2+ ions to Cu metal, releasing energy in the process. We can write the chemical reaction as:
$$\ce{Zn + Cu^{2+} -> Zn^{2+} + Cu} \tag{1}$$
Watch:
If we combine the two half-reactions shown earlier, we’ll see that the overall electrochemical reaction is the same:
$$\ce{Zn + Cu^{2+} <=> Zn^{2+} + Cu} \tag{2}$$
The overall standard potential for this reaction is the difference between the positive and negative standard electrode potentials, which for this case is 1.1 V.
This tells us that if we connect the terminals of the Daniell cell to an external circuit and discharge the battery, Zn will oxidise to form Zn2+ at the negative electrode, Cu2+ will reduce to form Cu at the positive electrode, and electrons will flow from negative to positive through the external circuit. If the Daniell cell contains Cu2+ and Zn2+ solutions with concentrations of 1 mol dm-3, and is at 25 °C and 1 atm pressure, then the voltage between the terminals will be 1.1 V, minus any energy losses (which we’ll get to later).
The Daniell cell is therefore operating as a galvanic cell, which is converting the stored energy of a spontaneous chemical reaction into electricity. However, we can also force current into the cell in the opposite direction; in this case, it would operate as an electrolytic cell, where energy is required to drive a non-spontaneous chemical reaction. This process would then recharge the battery, where its stored energy can be released in the galvanic reaction at a later time.
Nernst equation
Once we start passing current through the Daniell cell, we start moving away from standard conditions. One of the most fundamental equations in electrochemistry for calculating electrode potentials is the Nernst equation:
$$E = E^\plimsoll - \frac{RT}{nF}\ln Q \tag{3}$$
In this equation, the electrode potential
$E$ depends on the standard electrode potential
$E^\plimsoll$ and the reaction quotient,
$Q$. The reaction quotient is the product of the activities of the reaction products divided by the product of the activities of the reactants. We can write the Nernst equation for the complete Daniell cell like so:
By definition, the activities of the pure Zn and Cu metals are 1, and we can approximate the ratio of the activities of the Zn2+ and Cu2+ species by the ratio of their concentrations. We can therefore re-write the equation as:
If we plot the cell potential vs the concentration of Cu2+, we will see that the potential gradually decreases until we have almost completely depleted Cu2+ in the system:
With the Nernst equation, therefore, we can predict the discharge voltage curve for this battery system.
Selecting electrode materials for batteries
Tables of standard electrode potentials for a huge variety of redox couples can readily be found on many websites (e.g., here) or in textbooks. In theory, any combination of two redox couples may form the basis for a battery. For example, we can find from the table of standard potentials that lithium (Li) has an extremely low standard potential of -3.01 V vs SHE, which tells us it is an extremely potent reducing agent (i.e., it really wants to be Li+). Similarly, we can find that fluorine (F) has an extremely high standard potential of 2.88 V vs SHE, which indicates that the oxidised form, F2 gas, is a very potent oxidising agent (and really wants to be F-). In principle, this would tell us that a Li–F2 battery has a theoretical cell voltage in the neighbourhood of 5.89 V, much higher than the Daniell cell. However, these numbers on their own don’t tell us much else about the properties of this combination as a battery - its rate (power) capability, rechargeability, safety, and so on. We will learn more about these factors as we continue through this guide.
Get involved
Introduction to Li-ion Battery Chemistry
31 words (1 min) Matthew Lacey 28 Mar 2023
In Introduction to Li-ion Battery Chemistry we will introduce the fundamental operating principles of Li-ion batteries, from thermodynamics and kinetics, to material selection, and influence of physical phenomena on device performance.
Get involved
Subsections of Introduction to Li-ion Battery Chemistry
On this page we’ll take a look at the factors which determine electrode potential in intercalation electrode materials. We’ll look at the chemical potentials of the ion and the electron, and the lattice gas model: a basic model which helps us understand the origin of the different potential/voltage profile “shapes” we see in practice.
A (re-)introduction to intercalation materials
In our introduction to the thermodynamics of batteries we looked at the concept of the Nernst equation to understand what determines the electrode potential and ultimately the cell voltage. Here, we will look in more detail at the more complex chemistry which is the foundation of most Li-ion batteries, introduce a similarly fundamental model for understanding their electrode potential, and see how this helps us understand certain behaviours of these materials which we see in practice.
First, let’s remind ourselves of the basic construction of a Li-ion battery and the materials used. A schematic of a basic Li-ion cell is shown below. The arrows indicate the movement of ions and electrons when the cell is discharging.
A Li-ion battery is constructed from two electrodes, each made of a material capable of reversibly hosting and releasing Li+ ions; between them is a separator, which holds a (typically) liquid electrolyte which transfers those ions between the electrodes. The battery’s ability to store electricity relies on a large difference in the energy associated with hosting the Li+ ions in each electrode. That is, the removal of Li+ ions from the negative electrode and their insertion into the positive electrode should be accompanied by a large release of energy.
Chemical potential of lithium
Here, we will introduce the concept of chemical potential. The chemical potential is defined as the thermodynamic ability of a particle (e.g. an atom or ion) to perform physical work. It can be thought of as the energy required to introduce one more particle to the system, or alternatively the energy obtained by removing one particle from the system.
In an intercalation material, where the Li+ ion is incorporated into the structure of a host material, the electrode potential
$E$ is determined by the chemical potential of the lithium atoms in the structure
$\mu_\text{Li}$, which is in turn dependent on the chemical potential of the lithium ions and the chemical potential of the electrons in the structure. Mathematically, this is written as follows:
The units of chemical potential are typically kJ mol-1 and the units of Faraday’s constant are C mol-1. Dividing a chemical potential by Faraday’s constant, and accounting for a conversion of kJ to J, gives units for the potential of J C-1. One volt, 1 V = 1 J C-1.
Chemical potential of the electron
The chemical potential of the electron is equivalent to the Fermi level, and it is always negative. Broadly speaking, the chemical potential of the electron is largely determined by the redox-active constituents in the host material: the Fermi level is defined as the energy level at which we have a 50% probability of finding an electron. In transition metal oxides, for example, this is largely determined by the redox couple of the transition metal itself, similar to what we saw in the discussion of the Daniell cell. The atoms neighbouring the transition metal in the structure also play a pivotal role.
The schematic above illustrates the position of the Fermi level for some different materials. This illustrates the larger difference of the energy level of the Co3+/Co4+ couple vs Li/Li+ compared to Ti3+/Ti4+. It also illustrates the strong “inductive effect” of the PO43- anion in LiCoPO4 despite the lower oxidation states of the Co – but we will delve into this in more detail in a later page.
Chemical potential of the ion
A simple model for the chemical potential of the ion is the lattice gas model. This model assumes that when ions are introduced into the structure they are distributed randomly with no short-range order. We can call the resulting material a solid solution - the ions ‘dissolve’ completely into the host structure without significantly altering its structure.
In the lattice gas, the chemical potential of the ion changes with the fraction of occupied sites,
$x$, according to the following expression:
This right hand side of this equation consists of three parts.
$\mu_{\text{Li}^+}^\plimsoll$ is the standard chemical potential for the Li+ ion in the structure. This can be thought of as the “site energy”, which is primarily determined by electrostatic interactions with the neighbouring ions in the structure. The term
$RT \ln \left[ \frac{x}{1-x} \right]$ describes the contribution of the entropy of the ions in the solid solution, which is dependent on the fraction of the available sites in the structure which are occupied. Finally, we have a parameter,
$k$, for modelling non-ideal interactions between the ions in the structure.
$k$ is negative for attractive interactions.
We can plot this expression as a function of
$x$ and animate to show the effect of changing the non-ideal parameter
$k$. In the plot below, we have a y-axis of
$-\frac{\left(\mu_{\text{Li}^+}-\mu^\plimsoll_{\text{Li}^+}\right)}{F}$, or the change in the chemical potential of the ion relative to the site energy, divided by Faraday’s constant. The change in this value with ion concentration in the solid reflects the change we expect in the electrode potential.
As you can see in the plot, the change in the y-axis indicates that we expect a continuously decreasing electrode potential with increasing ion content in the electrode, with the steepest change closest to the extremes of fully empty or fully occupied sites.
The decrease in
$k$ (increase in ion-ion attraction) increases the slope of the curve. With a large enough
$k$, the slope increases such that the value of
$-\mu_{\text{Li}^+}$ begins increasing with increasing ion content, with a distinct minimum and maximum near
$x = 0$ and
$x = 1$. This is significant, because it predicts that with a strong enough ion-ion interaction, the material should phase-separate into two materials with high and low guest ion concentrations respectively, where the formation of distinct phases is more favourable than the solid solution. In this case, we have a “two phase” intercalation reaction.
Does this model hold in practice?
In commercially available Li-ion battery materials, no, not really – the materials we commonly use in batteries today behave significantly differently than the ideal behaviour, but the model is still useful for understanding some of the features of intercalation materials.
That said, there is at least one class of materials which does fit the model behaviour relatively well. Chevrel phases, which are a broad class of compounds with the general formula MxMo6X8, where X is either S or Se. These compounds form slightly distorted cubic structures with very large vacancies where the guest ion M can be incorporated and be able to diffuse in all three crystallographic directions, and freely due to the ‘softness’ of the anion. The vacancies in Chevrel phases are so large that they can readily incorporate relatively large ions such as Pb2+ or Ag+. The structure of one such compound, AgMo6S8, is shown below (data for the model can be found here).
A lithium Chevrel phase, LixMo6Se8 was studied in detail in the mid-1980s([1],[2]) and demonstrated to fit the lattice gas model very closely for the composition with a Li content of up to 1 Li per Mo6Se8 unit. The fit of the model also correctly predicted the phase separation of the material into two phases at lower temperature.
Supporting literature
[1] S.T. Coleman, W.R. McKinnon, J.R. Dahn, “Lithium intercalation in LixMo6Se8: A model mean-field lattice gas”, Phys. Rev. B29, 4147 (1984)
[2] J.R. Dahn, W.R. McKinnon, J.J. Murray, R.R. Haering, R.S. McMillan, A.H. Rivers-Bowerman, “Entropy of the intercalation compound LixMo6Se8 from calorimetry of electrochemical cells”, Phys. Rev. B32, 3316 (1985)
Get involved
Mass Transport: Transport and Transference
1018 words (5 min) Matthew Lacey 28 Mar 2023
Info
This content has been transferred from lacey.se and is not updated for this site yet.
Transport
The performance of a battery depends heavily on the properties of its electrolyte. Total ionic conductivity is one part of this, but how the current is carried by the electrolyte has some important implications, especially when the battery is subjected to very high charge or discharge currents.
Let’s consider the discharge of a Li-ion battery, containing an electrolyte with a simple salt such as LiPF6, and which is completely dissociated in the solvent. Current is drawn from the cell; Li+ ions are extracted from the negative electrode and inserted into the positive electrode. Part of this current is carried by the transport of Li+ ions, and the rest is carried by the transport of PF6- in the opposite direction:
We call the fraction of the current carried by a specific species, e.g., Li+ or PF6-, the transport number, with the symbol t+ or t_ respectively. The sum of the transport numbers is 1, and by this definition, any transport number for a specific species must be between 0 and 1 (we will revisit this later). A typical value for t+ in a Li-ion battery is usually calculated to be around 0.3.
What does this mean? The transport number is effectively a ratio of the mobilities of the ions, which, following the Nernst-Einstein equation (more on this later) also relates the partial conductivities and the diffusion coefficients to each other:
t+ is often smaller than t-, because the non-coordinating anion is often more free to move in the electrolyte, while the “hard” Li+ usually has to drag a large solvation shell with it, or is relatively tightly bound to a polymer electrolyte backbone.
Back to the battery. Because only a part of the current goes into moving Li+ ions, Li+ is consumed at the interface with the positive electrode (or produced at the negative) faster than it is replenished by electrical migration. This creates a gradient in salt concentration in the electrolyte, between the electrodes of the battery. The gradient in concentration then drives diffusion of the salt, which makes up for the rest of the transport of Li+ which isn’t supplied by migration.
The formation of this concentration gradient can ultimately limit the discharge (or charge) of the battery. If the concentration of salt at an electrode surface reaches zero, the ionic resistance becomes huge, and the battery stops. If the concentration of salt becomes too high, the salt can precipitate out altogether, and the resistance can again become huge. (These processes are, however, reversible, in time.)
The value of t+, and the salt diffusion coefficient, determine how fast this concentration gradient forms, and in turn the maximum current that the electrolyte can sustain indefinitely (assuming nothing else is limiting). Both of these parameters are therefore important properties of any electrolyte, in addition to its total ionic conductivity. Ideally, t+ = 1 (and, correspondingly, t- = 0 - implying total immobilisation of the anion). In this situation, a concentration gradient cannot form at all.
Transference, not to be confused with transport
Well, one important consideration, regardless of how carefully the experiment is set up, is that assumption of adherence to the Nernst-Einstein equation. This equation assumes that the ions do not interact with each other when they are dissolved, but this is only even approximately true in very dilute solution, for example concentrations < 0.01 M.
At typical salt concentrations used in batteries - 1 M, or maybe even higher - there are significant interactions between ions. Take a generic salt, LiX, for example. When we dissolve this into a solvent at a relatively high concentration, this dissociates into solvated Li+ and X- ions, but some will remain as neutral [LiX] ion pairs.
We can also consider the potential formation of ion triplets such as [Li2X]+ and [LiX2]-. Whether or not such species really exist is besides the point, but in considering these examples it is clear that the migration of the [Li2X]+ with only 1 positive charge moves two Li+; and the migration of [LiX2]- moves a Li+ in the opposite direction! These each have their own transport numbers. In this case, we should consider the concept of transference, distinct from transport. This is, in fact, the more important concept when it comes to limitations in the battery itself.
The transference number for lithium, for example, is defined as the number of moles of lithium transferred by migration per Faraday of charge. To avoid confusion with the transport number, we will use the symbol T+ (and the transference number of the anion, correspondingly, is T-). It is important to know that transference and transport are often used interchangeably, and usually with the lower case symbol t. However, they are not the same. From the above examples, it should be clear that:
T+ therefore quantifies the net transference of all the Li+-containing species which migrate in the electrolyte. Of course, in an ideal system, where there is no ion association, then T+ = t+.
As with the transport numbers:
$$T_+ + T_- = 1$$
However, there are no bounds on individual transference numbers. From the equation above, it is theoretically possible to have T+ < 0 if the mobility of the [LiX2]- triplet is such that it transfers Li+ in the “wrong” direction faster than the other species transfer Li+ in the “right” direction.
That’s not all though: while this is all happening, neutral [LiX] ion pairs - which do not migrate, because they are neutral - are diffusing down the concentration gradient and also, in effect, transferring Li+. However, this is not included in the definition of T+, because the definition of T+ considers only migrating species.
Ultimately, a system could have T+ < 0 and still operate, but diffusion of neutral ion pairs would have to be fast enough to both supply the current passed and compensate for the net effect of migration to transfer Li+away from where it should be.
Get involved
Chapter 2
Battery Materials
In Materials we will provide an introduction to the chemistry of specific battery materials, beginning with the major commercialised families and introducing the development of new materials.
The Current materials section will introduce the materials that comprise current battery technology.
Get involved
Subsections of Current materials
Separators
327 words (2 min) Marcel Drüschler and Matthew Lacey 05 Aug 2023
Info
This page is incomplete. If you’re interested in contributing, please check our About page, information for interested contributors and get in touch!
Let us focus on an important component of Li-ion batteries: the separator.
The separator is a very thin porous membrane that is made of an electrically insulating material and prevents the electrodes from being in direct contact with each other. Important factors that influence the performance of a separator as part of the Li-ion battery are:
porosity,
pore size and pore distribution, which also affects the tortuosity,
the material the separator is made of, in particular its electrical and mechanical properties, and
the thickness.
Requirements
There are a number of requirements that a separator material must meet for practical application.
Technical requirements
The separator material must fulfil a number of technical requirements. The separator:
should allow a good wettability for the chosen electrolyte solution,
must be chemically inert in contact with the electrolyte solution and electrode reactants,
should be mechanically and dimensionally stable and tolerate the temperature window that is given by the final application,
should be an electronic insulator,
should minimise the loss of effective conductivity to maximise power density,
should be thin and light to maximise energy density.
Economic requirements
The separator material:
should be commercially available,
should be available from several sources,
should be available at a reasonable price.
Ecological requirementss
The separator material:
should ideally be recycleable,
should be accessible by an energy-efficient synthesis strategy.
Common materials
Battery separators in commercial use are most typically porous poly(ethylene) or poly(propylene). Thin coatings (ca. 2 µm) of aluminium oxide (Al2O3) or boehmite (AlO(OH)) particles on one or both sides are common, to enhance mechanical, thermal and electrochemical stability.
In Experimental Electrochemistry we will cover the experimental electrochemical methods used in battery research and development, beginning with the fundamentals of experimental design and instrumentation.
This introduction to electrochemical impedance spectroscopy (EIS) began in 2016 as a result of what I saw as a lack of easily accessible and useful information for learning the principles, theory and practical application of EIS for most users (that is, non-electrochemists). The way I saw it, there was very little help available for those who got the basic idea of EIS but were struggling to fit their data or weren’t sure how to set up their experiments. If you’re looking to get a handle on EIS but aren’t sure where to start, these pages are, hopefully, for you.
The pages that follow include basic explanations starting from the beginning, along with some simulation web apps and some exercises where you can get a feel for working with impedance data. You can get started by following the links below, or in the main menu. Comments and suggestions for the Content Roadmap are always very welcome.
First off, I want to say that although I’ve been a user of impedance spectroscopy since 2008, on an applied level (battery characterisation) it still confuses the hell out of me on a regular basis. Learning the theory is one thing, but in-depth analysis, especially when it comes to equivalent circuit fitting, is hard – it requires a healthy degree of intuition, and usually needs very carefully designed experiments to get meaningful results… but sometimes it’s just a pain. I’d dare to suggest that a large proportion - maybe even a majority - of the people in the battery field who have done some impedance spectroscopy work in peer-reviewed publications do not really understand what they’re doing with it.
But, don’t be discouraged(!). It’s not easy to learn, but in writing this I’ve done my best to keep the level as accessible as possible, to give examples, some little apps where you can do simulations to see how changing parameters in a model system changes things, and many more, to try and give you the best chance to build up that level of intuition.
I used to finish my lectures with “Matt’s top tips for impedance spectroscopy”, to try to restore some of the enthusiasm and confidence I’ve just stolen from my poor students, and here they are:
Matt’s top tips for impedance spectroscopy
EIS can be ambiguous, and a good equivalent circuit fit can be elusive (but don’t let that put you off)
The fewer parameters an equivalent circuit has, the better
Always check the fitted values make sense (and the errors are reasonable).
Practice makes perfect!
Download [some simulation/fitting software of your choice] and play around*…
* An example is ZView (Scribner Associates). Older versions (e.g. 3.2) allow you to do simulations as well as equivalent circuit fitting of data. Perhaps Google can help you out…
Get involved
Subsections of Electrochemical Impedance Spectroscopy (EIS)
Principles of EIS
1231 words (6 min) Matthew Lacey 15 Feb 2023
Defence against the dark arts
Since you’re reading this, you most likely know that as the name suggests, Electrochemical Impedance Spectroscopy (or just EIS, from now on) involves looking at the impedance characteristics of an electrochemical system over a range of frequencies (that’ll be the spectrum part). And maybe you’re thinking about trying your hand at some equivalent circuit fitting, which is the dark art process of fitting a spectrum you’ve measured to a model based on some real (and some not real) electrical components. But first, it’s important to understand some of the theory behind impedance itself.
Impedance vs resistance
Consider Ohm’s law, which describes the relationship of voltage to a direct current passing through a resistor:
$$E = IR \tag{1}$$
Impedance is, very simply, extends the concept of resistance to an alternating current, and generally represented as
$\pmb{Z}$. So you can think of it, simply, like this:
$$E = I\pmb{Z} \tag{2}$$
We’ll come back to this in a moment. For now, it should be clear that a measurement of impedance, therefore, can be made by made simply by applying an oscillating voltage, and measuring the (oscillating) current response. We can write down an equation for the oscillating voltage we apply like so:
$$E(t) = \left|E\right|\sin(\omega t) \tag{3}$$
where
$\left|E\right|$ is the amplitude of the voltage signal, and
$\omega = 2 \pi f$ (the angular frequency). The response will be a current with an amplitude
$\left|I\right|$, which is also shifted in phase from the applied signal:
$$I(t) = \left|I\right|\sin(\omega t + \theta) \tag{4}$$
The current is shifted in phase because of reactance (e.g., a capacitance or inductance) in addition to the resistance (which changes the amplitude). The impedance can therefore be expressed like this:
$$\pmb{Z} = \frac{E(t)}{I(t)} = \frac{|E| \sin(\omega t)}{|I| \sin(\omega t + \theta)} = |Z| \frac{\sin(\omega t)}{\sin(\omega t + \theta)} \tag{5}$$
Have a look at the animation below. The ‘current’, I, is 72° out of phase with the ‘voltage’. The graph on the right is known as a Lissajous curve, showing the relationship between I and E. In the past, impedance spectroscopy was done by obtaining these curves on an oscilloscope and analysing them. Thankfully, it’s all a bit easier nowadays.
Impedance as a complex number
Ok, complex maths time. Without going into too much detail, via Euler’s formula:
$$e^{jx} = \cos(x) + j \sin(x) \tag{6}$$
we can re-write all of the above using complex numbers:
$$\pmb{E} = I \pmb{Z} = I\left|Z\right|e^{j \theta} \tag{8}$$
Note that
$j$ is the imaginary unit, i.e.,
$j = \sqrt{-1}$, which we use instead of
$i$ to avoid confusion with the symbol for electrical current. You can see from the above equation that the ratio of an oscillating voltage to an oscillating current is the impedance, which has a magnitude
$|Z|$ and a phase angle
$\theta$. You can think of this as a polar coordinate representation. More commonly for impedance spectroscopy, however, we generally use the Cartesian complex plane representation, dividing the complex impedance into the real and imaginary parts:
$$\pmb{Z} = Z' + j Z'' \tag{9}$$
$Z'$ and
$Z''$ are the resistive and reactive parts of the impedance respectively. You’ll see this more clearly on the page about the impedance of simple RC circuits.
We can represent any
$\pmb{Z}$ on an Argand diagram, as in the graph below. This is the basis for the Nyquist plot, which is the plot of the real and imaginary parts of the impedance that you’ll come across most often. An impedance measurement for a single frequency is a single point on a Nyquist plot. An impedance spectrum is therefore a series of points, where each point is a different frequency.
These plots are visually useful, because the characteristic shapes that can appear in the plots as you’ll see later can give you a rough idea of what you’re looking at. The downside, though, is that you can’t know what the frequency associated with a particular point is from looking at the Nyquist plot alone, and so the plot doesn’t contain all the information you need. This is why the alternative Bode plot – plots of
$\log Z'$ and
$\log Z''$ vs
$\log f$, or
$\log |Z|$ and
$\theta$ vs
$\log f$ – are still important.
Nyquist plot
I’ll finish up this page by briefly introducing a typical Nyquist representation of an impedance spectrum itself. The plot below is data I acquired from a Li-ion test battery, and fitted to a model myself. The frequency range the points represents is between 100 kHz and 100 mHz. This is fairly typical for most systems, although depending on what you want to measure you might go up to 1 MHz or more, or as low as 1 mHz. So how do you make sense of this plot? Well, there are three things I’ll note for now.
First, the impedance is always lowest (i.e., smallest values of
$|Z|$ at the highest frequency, so you can see that the frequency decreases if we follow the curve from the points near the origin to the points in the top-right corner. Secondly, you’ll note (as in the Argand diagram above) that the values of
$Z''$ are negative (plotted as
$-Z''$). This will become clearer later, but by convention capacitance is a negative reactance, so impedance spectra will in most cases only have positive
$Z’$ values and negative
$Z''$ values.
Lastly, you’ll note the shape of the spectrum, particularly the semi-circle part. The shapes you see in the Nyquist plots can be characteristic of certain elements or combinations of elements, so they are (often, but not always) visually useful for quickly understanding something about the system you’re measuring. Because of this I was able to take this relatively good quality data, think of a reasonable model, guess a few of the parameters and then fit the entire spectrum relatively quickly. In the following pages you’ll read about the experimental technique I used to get this data as well as the elements of the model I’ve fitted the data to, and hopefully you’ll be able to see how it all fits together.
Square the axes!
There’s a slightly nitpicky but formal stylistic point I like to make when I give lectures on this topic, and that’s about axis scaling on Nyquist plots. As you can see from my Argand diagram above, the axes are proportional (i.e., 2 units on the x-axis are equal in length to 2 units on the y-axis). Formally, it should be this way, because you can still read any point as a magnitude and a phase angle as well as the real and imaginary parts. Practically, it is still helpful to do this because it will let you see important features such as 45° lines easily. Unfortunately, most people who publish impedance results in the literature do not do this. To some extent, I sympathise, because common scientific graphing programs like Origin Pro do not make it easy to do this. But, most often the person making the graph goes to the effort to set the limits on the x- and y-axes to be the same, but keeps the plot itself the default rectangle, rather than resizing it to a square. This, I think, is just sloppy.
And as a final tip for R and ggplot2 users: it is thankfully trivial to ensure that axes are kept proportional. Just add coord_fixed to ggplot()!
Get involved
RC circuits
1161 words (6 min) Matthew Lacey 15 Feb 2023
Overview
Now that we’ve introduced some of the maths behind impedance, and the plots we use to represent it, we can start to look at the mathematical definitions and the impedance response of two of the most basic electrical circuit components – resistors and capacitors – and combinations of them.
Impedance of a resistor
This is the simplest electrical circuit, and the easiest to understand. Resistors, as you surely know, obey Ohm’s law, so the current is always proportional to the voltage, there is no reactive part (i.e., phase shift) and so no dependence on frequency whatsoever. We can just write that down like this:
$$\pmb{Z}_R = R \tag{1}$$
where R is the resistance. The Nyquist plot for a resistor then is very simple – it’s just a single point on the x-axis at any frequency. The below plot shows the Nyquist plot for a resistor with a resistance of 20 Ω.
Impedance of a capacitor
Capacitors have a purely reactive impedance. An ideal capacitor has zero resistance. When an alternating voltage is applied across a capacitor, the current leads the voltage (the phase is -90°), and the impedance is inversely proportional to the frequency. That is, the impedance increases with decreasing frequency. Consider applying a DC voltage across a capacitor – after a long enough time, the capacitor is fully charged and no more current flows. The impedance is effectively infinity. The equation describing a capacitor is:
$$\pmb{Z}_C = \frac{1}{j \omega C} \tag{2}$$
where
$j$ is the imaginary unit,
$\omega = 2 \pi f$ and
$C$ is the capacitance.
he Nyquist plot for a capacitor therefore looks like a vertical line, where Z’ = 0 for all frequencies. The below plot shows the Nyquist plot for a capacitor with a capacitance of 1 mF, in the frequency range 1 kHz – 10 Hz. The highest frequency points have the lowest impedance, with the impedance increasing as frequency decreases.
Capacitances arise all over the place in electrochemical systems, pretty much anywhere you have an interface – most often from the capacitance of the double layer, but also dielectric capacitance, or at grain boundaries in solids, and so on.
This is all fairly straightforward so far, so now we’re going to consider combining some of these different circuits together.
RC circuits
Series RC circuit
In series, the impedances are additive:
$$\pmb{Z} = \sum_n \pmb{Z}_n \tag{3}$$
The impedance of the series RC circuit is therefore just the addition of the individual impedances of the resistor and the capacitor together:
The Nyquist plot for a series RC circuit (where R = 5 Ω, C = 1 mF, in the frequency range 1 kHz – 10 Hz) is shown below. As you might expect, the real impedance Z’ is equal to the resistance of the resistor for all frequencies, and the imaginary part of the impedance follows the same behaviour as for the ideal capacitor.
You can consider the series RC circuit as a simple model for things like a blocking interface – for example an inert electrode immersed in a conducting electrolyte – where R represents the ionic resistance of the electrolyte, and C represents the capacitance of the double layer on the electrode surface.
This also represents two electrodes in an electrolyte (i.e., a complete cell), because a series circuit with two capacitors (i.e., C-R-C) simplifies down to a single RC unit anyway, if you follow the equations above.
Parallel RC circuit
Now, let’s consider the parallel case.
In parallel, the admittances (i.e., the reciprocals of the impedances) are additive:
So then we can write the expression for the parallel RC circuit like so:
$$\frac{1}{\pmb{Z}} = \frac{1}{R} + j \omega C \tag{6}$$
If we rearrange that equation for
$\pmb{Z}$ (by first multiplying all the terms by R) then we end up with:
$$\pmb{Z} = \frac{R}{1 + j \omega RC} \tag{7}$$
From this equation you can see that at high frequency, i.e.,
$\omega \rightarrow \infty$, the lower term on the fraction goes to infinity, so the impedance tends towards zero; the ideal circuit behaves like the capacitor at infinite frequency – it has zero impedance. At low frequency, i.e.,
$\omega \rightarrow 0$, however, you can see that the bottom term becomes 1, so the total impedance of the circuit equals R – i.e., with a direct current, the circuit behaves like a resistor (this makes sense, right? Eventually with a direct current, the capacitor becomes fully charged and the current only goes through the resistor). The Nyquist plot for this circuit, then, is a semicircle, intercepting the real (Z’) axis at 0 and R. The below plot shows the Nyquist plot for a parallel RC circuit where R = 5 Ω, C = 1 mF, in the frequency range 1 kHz – 1 Hz:
Semicircles in the Nyquist plot are very common in electrochemical impedance, and are usually associated with processes such as charge transfer, because at an electrode surface the transfer of charge happens in parallel with the charging of the double layer capacitance – hence the semicircle.
You’ll also note that I’ve marked the very top of the semicircle with f*. This is known as the relaxation frequency, and relates to the RC time constant of the circuit. From the previous equation, you will see that the peak of the semicircle occurs when
$\omega RC = 1$. The time constant is then defined as follows:
This is an important concept in EIS, because it tells us something about the timescales on which different processes are occurring. This equation also allows you to calculate capacitances in these elements knowing only the resistance (from the diameter) and the relaxation frequency.
This is where the Bode plot comes in handy. The top of the semicircle simply appears as a peak in a plot of the imaginary part against the frequency, with log scaling:
Since I simulated the circuit with values of R = 5 Ω and C = 1 mF, you should be able to follow that:
and see on the Bode plot above that the peak is at around 32 Hz.
Another reason this is important is that the time constant (that is, the frequency dependence) of these elements of course affects the order in which they appear in the Nyquist plot. Elements with a smaller time constant (i.e., a higher relaxation frequency) will, naturally, appear at higher frequencies in the impedance spectrum. Of greater concern, however, is that elements with very similar or the same time constants will tend to overlap. I’ve included a Shiny app for simulating a circuit made up of two parallel RC units in series to show this. Have a go at changing the parameters, and see how they affect what the Nyquist plot looks like.
Get involved
Multiple Time Constants
537 words (3 min) Matthew Lacey 28 Mar 2023
Overview
Here, we’ll look at the series combination of two parallel RC circuits, and in particular how the relaxation time constants influence our interpretation of the results.
Let’s consider the impedance of this circuit:
If you have familiarised yourself with the contents of the previous page you should recognise that we can calculate the impedance of this circuit, which we can refer to as an RC-RC circuit, by this equation:
You might also see from this equation that we expect the response of this RC-RC to be two semicircles, one for each separate parallel RC ‘unit’, each with their own RC time constants. But how do we know which semicircle corresponds to which parallel RC ‘unit’?
The critical thing to understand is that, in a Nyquist plot, the semicircles appear in order of increasing time constant (and, in turn, decreasing relaxation frequency). Parallel RCs with a smaller RC time constant (smaller
$R \times C$ product) will appear first, at higher frequencies, and higher time constant processes will appear later, at lower frequencies.
To illustrate this, we can consider the response of a hypothetical circuit as illustrated above, with fixed resistors but capacitors of different values, to vary the RC time constants. In the below example, we have R1 = 5 Ω and R2 = 15 Ω, and the following three cases where τ1 = R1C1 and τ2 = R2C2:
C1 = 10-5 F, C2 = 10-3 F; therefore τ1 < τ2
C1 = 10-3 F, C2 = 10-5 F; therefore τ1 > τ2
C1 = 10-4 F, C2 = 3.3 × 10-3 F; therefore τ1 = τ2
The Nyquist plots of these cases is given below:
As you can see, in the first case the 5 Ω semicircle appears on the left of the Nyquist due to its lower time constant. In the second case, swapping the parallel connected capacitors over changes the time constant, so that the 5 Ω semicircle has a larger time constant, has therefore a lower relaxation frequency, and instead appears on the right of the Nyquist plot.
In the third case, the capacitances have been selected so that the time constants are equal. In this case, the two capacitors charge at exactly the same rate, and the response of the circuit will be identical to that of a single parallel RC circuit, with a total resistance at zero frequency (DC) of R1 + R2.
This last case has significant practical implications, as it tells us that if we have two separate processes with very different associated R and C values, if they have similar or equal time constants (i.e., they occur on a similar time scale), then they may be extremely difficult (or impossible) to separate. This is a fundamental challenge with EIS.
We can also visualise this with the below animated plot (kindly provided by Dr Sam Cooper). The below plot similarly shows an R-RC-RC circuit where one capacitor value is varied (all other components fixed) to change the time constant, and the order in which they appear. You will see that the closer the two time constants are to each other, the more they overlap.
Get involved
Constant Phase Elements
601 words (3 min) Matthew Lacey 28 Mar 2023
Overview
So far, we’ve looked at the impedance response for some ideal resistors and capacitors and some simple combinations of them. Unfortunately, in electrochemical systems we often encounter processes which don’t have a “real” equivalent electrical component, so we have to invent some. One of the most common such elements is the constant phase element, or CPE.
One of the most common circuit elements for modelling non-ideal behaviour is the constant phase element (CPE, or sometimes Q). This is a common symbol for this circuit element:
You might think that it looks like a wonky capacitor – and you’d be right, because this circuit element exists largely to describe capacitance as it appears in real electrochemical systems, because of things like rough surfaces, or a distribution of reaction rates. It is an imperfect capacitance – the effective capacitance and ‘real’ resistance are increasing as the frequency decreases. The origins of CPE behaviour are numerous and some of them quite complex, but there are good guides explaining this elsewhere.
The mathematical definition is very similar to that of the capacitor as well:
$$\pmb{Z}_Q = \frac{1}{Q_0 (j \omega)^n}$$
where n is the constant phase,
$(-90 \times n)$°, and n is a number between 0 and 1. Be aware though – this is not the only possible definition of the constant phase element – it can also be defined by putting the Q0 value inside the brackets.
So, what does the Nyquist plot look like? You might have guessed by now that the Nyquist plot for a CPE looks similar to a capacitor – a straight line, but with a phase of
$(-90 \times n)$°. The below plot shows the Nyquist plot for a CPE with a Q of 1 mS sn, where n = 0.85 (more on those units later), in the frequency range 1 kHz - 10 Hz.
R-CPE or RQ circuits
It should be fairly clear by now what the Nyquist plot of a series R-CPE element looks like, so I’ll only show the parallel case, the characteristic “depressed” semicircle. The below plot shows the Nyquist plot for a CPE with a Q of 1 mS sn, where n = 0.85, in parallel with a 5 Ω resistor, in the frequency range 10 kHz - 10 Hz.
The Bode plot for a parallel R-CPE circuit looks very similar to that of a parallel RC circuit, but with a subtle difference: on a log-log plot (i.e., log(-Z’’) vs log f), the gradient of the straight line gives you the n value:
You may be able to see from the above plot that in the high frequency region (to the right of the peak) the gradient is -0.85, and in the low frequency region (to the left of the peak) the gradient is +0.85. Being able to estimate these values from Bode plots can be a useful technique to estimate parameters when performing equivalent circuit fitting, as we will see later on in this section.
Can I calculate the real capacitance?
Q0, according to the mathematical definition of the CPE, has units of S sn (that’s siemens-seconds-to-the-power-n), which have no real physical meaning. However, it is possible to determine the actual capacitance behind the CPE when you have a parallel R-CPE circuit. Think about the units again – you should be able to see that:
$$RQ_0 = \tau^n = (RC)^n$$
Rearranging this equation gives:
$$C = \frac{(RQ)^{\frac{1}{n}}}{R}$$
This equation holds as long as the phase angle does not deviate too far from -90° (n > 0.75). I won’t go into this further, though – the ConsultRSR webpages on EIS provide some useful reading on this point.
Get involved
Diffusion Impedance
1131 words (6 min) Matthew Lacey 28 Mar 2023
Overview
Mass transport processes, such as diffusion, migration and convection, are a key aspect of electrochemical systems. In this subsection, we will look at fundamental models for the effect of diffusion processes on impedance. As with the CPE, we invent some new circuit elements to describe these, but these can be approximated using the familiar resistors and capacitors, as we will see.
Semi-infinite diffusion
The simplest and most common circuit element for modelling diffusion behaviour is the Warburg impedance or Warburg element, which models semi-infinite linear diffusion – that is, diffusion in one dimension which is only bounded by a large planar electrode on one side.
The equation for this element is relatively simple:
where
$\sigma$ is the Warburg coefficient, with units of Ω s-1/2 – we’ll come back to this shortly. In the Nyquist plot, the Warburg impedance gives a straight line with a phase of 45°, which is very recognisable in EIS. If you spot a 45° line in the Nyquist plot, it is usually associated with diffusion. The below plot shows the Nyquist plot for a Warburg element with
$\sigma$ of 10 Ω s-1/2 in the frequency range 10 kHz - 10 Hz.
And if you’re looking at this thinking that this line look very much like a constant phase element but with a phase angle of 45°, then you’d be right – mathematically they are very much the same. In fact, some popular impedance analysis softwares do not actually provide a semi-infinite Warburg element, so a convenient alternative for this is a CPE element with n fixed at 0.5. This gives slightly different numbers back when fitting – if you use a CPE, you will get back the Q0 value, rather than the Warburg coefficient
$\sigma$. The two values are related though, by the equation:
$$\sigma = \frac{1}{\sqrt{2} \cdot Q_0} $$
But what does
$\sigma$ actually mean? This is somewhat more complicated, but if you consider the case where you have a soluble redox couple in solution, with a nice reversible electrochemical reaction like:
$$\text{O + n e}^- \rightleftharpoons \text{ R} $$
then
$\sigma$ is related to the diffusion coefficients and concentrations of those species:
here, in addition to the usual constants,
$D_O$ and
$D_R$ are the diffusion coefficients and
$c^\infty_O$ and
$c^\infty_R$ are the bulk concentrations for the species O and R respectively.
The transmission line
Conventional “Fickian” diffusion is not the only process which gives rise to this type of impedance in electrochemical systems. In batteries, the porosity of electrodes also gives rise to a similarly characteristic 45° line in the Nyquist plot.
This was described in detail by de Levie, who proposed the transmission line model for an electrode with cylindrical pores filled with electrolyte:
Consider the two parallel “rails” as being the electronic resistance in the electrode material itself and the ionic resistance in the electrolyte respectively. The capacitors represent the double layer capacitance.
This equivalent circuit, when infinitely long, gives an impedance response which is identical to the Warburg impedance above. Practically, however, porous electrodes have a finite length, and so show a 45° line only in a certain frequency range. The impedance response due to finite diffusion is discussed below.
A final point on the transmission line: if the transmission line shows the same response as Fickian diffusion, can the case of a porous electrode be considered diffusion as well? In a sense, yes. The movement of ions through the pores is coupled to the movement of electrons through the pore walls. This is an example of ambipolar diffusion.
Finite diffusion
Often in the “classic” electrochemical setups, diffusion often appears semi-infinite because the timescale of the experiment is not long enough for the system to reach a steady state. However, in many real systems and in some standard experiments, diffusion is either naturally, or by design, limited. This gives rise to finite diffusion behaviour, which shows a different response than the standard Warburg impedance.
There are two important equivalent circuit elements for finite diffusion. They are the finite length Warburg (FLW) and the finite space Warburg (FSW), sometimes called the “short” and “open” Warburg elements respectively. Their responses in a Nyquist plot look like this, with parameters Z0 = 10 Ω and
$\tau$ = 0.075 s (more on these parameters shortly) in the frequency range 10 kHz to 10 Hz:
Let’s look at the FLW first. Mathematically, it can be written as:
The two parameters
$Z_0$ and
$\tau$ reflect the properties of the system or the process giving rise to the FLW behaviour. The impedance of the FLW tends to the value of
$Z_0$ at low frequency. At high frequencies, the response is almost exactly that of the Warburg impedance. The shape of the FLW in the Nyquist plot therefore looks like a 45° line at higher frequencies, and transitions into a semi-circle shape at low frequencies. It can be thought of as a Warburg being “shorted” by a resistor - although a parallel W-R circuit will not give the same response. It can also be modelled by a finite length transmission line which is short-circuited at one end. For this reason it is sometimes also known as a “short Warburg”.
This response is typically associated with diffusion (or more generally mass transport) through a layer with a finite length. A classic example of this is the response of the rotating disk electrode - the point of which being to reduce the distance from the electrode to the bulk by controlling convection, rather than diffusion in this case.
In this case, the response tends towards capacitive-like behaviour at low frequencies, where
$Z’(\omega = \infty) = Z_0/3$. This response is typically associated with diffusion where one of the boundaries is blocking, such as in a porous electrode, as discussed previously on this page. This response is also associated with the diffusion of ions within a storage electrode, such as in lithium-ion batteries. This element can also be modelled as a finite length transmission line which is “open” at one end, and for this reason is sometimes also known as an “open Warburg”.
Mind the definitions!
You can find a number of different definitions of these finite elements and it’s easy to get confused. One source of confusion can be from defining these elements in terms of admittance. For example, the FLW is frequently defined like this:
You might have noticed that this definition contains
$\coth$ instead of
$\tanh$. In this case it’s important to remember that:
$$\coth(x) = \frac{1}{\tanh(x)}$$
Get involved
The Randles Circuit
559 words (3 min) Matthew Lacey 28 Mar 2023
Overview
The Randles circuit is the probably the most recognisable equivalent circuit model in the world of electrochemistry and electrochemical impedance spectroscopy. We’ll look at what processes it models, why the model is arranged in the way it is, and its distinctive Nyquist plot.
The Randles circuit, given below, is a model for a semi-infinite diffusion-controlled faradaic reaction to a planar electrode.
That’s a lot of technical terms, I know. But the Randles circuit has plenty of significance to real electrochemical reactions, and if you’ve already worked your way through the previous pages in this section you should now be familiar with all of the elements.
A simple model for an electrode immersed in an electrolyte is simply the series combination of the ionic resistance, Ri, with the double layer capacitance, Cdl. If a faradaic reaction is taking place, of the sort:
then that reaction is occurring in parallel with the charging of the double layer – so the charge transfer resistance, Rct, associated with the faradaic reaction is in parallel with Cdl.
The key assumption is that the rate of the faradaic reaction is controlled by diffusion of the reactants to the electrode surface. The diffusional resistance element (the Warburg impedance, W), is therefore in series with Rct.
And that’s all there is to it. You will see impedance responses with this sort of shape in all manner of electrochemical systems, although often with multiple semicircles and other overlapping processes.
Nyquist and Bode plots
A typical Nyquist plot for the response of a Randles circuit is shown below. The distinctive shape of the Nyquist plot for the Randles circuit is the semicircle created by the Ri-(RctCdl) portion of the circuit, followed by the 45° line characteristic of the Warburg impedance. These two separate parts of the circuit are indicated by the dotted lines on the plot.
The Bode plot for the same circuit follows below. As you will have seen on previous pages, we can similarly extract the relaxation frequency for the RctCdl time constant from the peak in Z’’ in the Bode plot.
Parameters for both of these plots: Ri = 1 Ω, Rct = 4 Ω, Cdl = 10-5 F, σ = 10 Ω s-1/2, in the range 1 MHz - 1 Hz.
A note on series and parallel combinations
The question of whether elements corresponding to physical processes should be in series or in parallel in a circuit model can often cause confusion. An important aspect to consider is whether the current which is associated with each element either adds to the currents from other processes, or limits (or is limited) by them.
In the case of the Randles circuit, the faradaic current (which flows through the Rct resistor) and the capacitive current (which flows through the Cdl element) are independent of each other, and as a result are additive:
The currents flowing in circuit elements which are in parallel add together to give the total current in the circuit, so physical processes which independently contribute to the total current are typically represented in parallel.
Conversely, if two processes act as a bottleneck for each other, the same current flows through both elements and they are therefore usually represented in series. In series, as we have seen earlier, the impedances of each element are additive.
Get involved
Debye Circuit
509 words (3 min) Matthew Lacey 28 Mar 2023
Overview
The Debye circuit is a common model used for determining ionic conductivity in polymer electrolytes. However, it can occasionally cause confusion and misinterpretation of results as the visibility of some processes in the impedance spectrum can depend on the material properties and experimental conditions. We’ll take a look at why this might be on this page.
The Debye circuit is often used to interpret the impedance spectra of polymer electrolyte films sandwiched between blocking electrodes (i.e., electrodes that don’t undergo any reaction themselves), such as steel, or gold. This is a common method for calculating ionic conductivity of these materials. The circuit looks like this:
where Ri is the ionic resistance, Cdl is the double layer capacitance (represented by a constant phase element), and Cd is the dielectric capacitance.
It is important to account for the dielectric capacitance when making these measurements, because of the small inter-electrode distance (giving a higher dielectric capacitance) and a much higher ionic resistance of a polymer electrolyte than a liquid electrolyte (often 3 orders of magnitude higher, or more).
You should now start to see how the relaxation frequency associated with the semicircle that arises from the parallel Ri-Cd combination might well appear in the high frequency (1 kHz – 1 MHz) part of an impedance measurement, where it would normally be well outside the measurable range if the electrolyte was a more conductive liquid. Failure to account for this can lead the experimentalist to mistakenly assign the ionic resistance to the highest-frequency intercept of the real axis and the lower frequency resistance to some unknown charge transfer resistance (it does happen, I’m afraid).
Visualisation
We can simulate the response of the Debye circuit for a typical polymer electrolyte between two blocking electrodes. Instead of directly using R and C values, we can calculate them from typical values of thickness, conductivity, and dielectric constant. If we say for example that we have a thickness
$l$ = 300 µm (thick, but typical for a lab experiment), dielectric constant
$\varepsilon_r$ = 10, and we allow the conductivity to vary between 1 and 0.001 mS cm-1 with a fixed non-ideal double layer capacitance of ~10 µF cm-2, we can expect the resulting Nyquist plots in the range 1 Hz - 1 MHz to look something like the below:
As the conductivity decreases, the resistance R increases, increasing the time constant R-Cd. It can be seen that that until the conductivity is below 0.02 mS cm-1, the semicircle associated with R-Cd is hardly visible as the relaxation frequency is well outside the measurement range. This hopefully shows more clearly why this phenomenon is rarely spotted away from measurements on poorly conductive samples. Under these conditions, the value of the capacitor Cd is ~4.4 × 10-11 F cm-1, which is over five orders of magnitude lower than the double layer capacitance and thus easily distinguished in an equivalent circuit fitting.
Some typical conductivities of different materials as a guide: polymer electrolyte: 10-6 S cm-1; Li-ion battery electrolyte: 4 x 10-3 S cm-1; aqueous electrolyte: 10-1 S cm-1.
Get involved
The Kramers-Kronig transform
367 words (2 min) Matthew Lacey 28 Mar 2023
I will not go into too much detail about the Kramers-Kronig (or, just K-K) transform other than to say that this is a method for validating impedance data, to give you some idea of whether it’s actually possible to fit an equivalent circuit. This is the equation:
Really, you don’t actually need to know this equation to do a K-K transform, because it is sometimes included as a function in the software for the instrument – but it is useful to know where it comes from. The Kramers-Kronig relations say – in this context – that you can calculate the imaginary part of the impedance from the real part of the impedance, and vice versa, provided the measured impedance response satisfies these conditions:
it’s causal – that is, the response only comes from the applied perturbation
it’s linear – that is, the response is first order (i.e., response does not change with larger perturbations). Unfortunately, electrochemical systems are almost often not linear – current changes non-linearly with applied voltage – so applied voltages are kept to as low an amplitude as possible (say, a few mV), to keep the response as linear as possible.
it’s stable – that is, the system does not change with time, or continue oscillating after the perturbation is removed.
it’s finite for all frequencies – this is an important one, because it says that the K-K transform cannot be applied to impedance spectra which are capacitive at low frequency, because the impedance tends towards infinity.
A response which does not satisfy all of these conditions probably cannot be fitted to an (appropriate) equivalent circuit.
A simpler alternative
Actually, there is an easier way you can check the validity of the impedance response without having to look for a K-K transform function. Typically, when running an impedance measurement, you will likely run through the frequencies from highest to lowest. Do that, but then run the measurement again with the order reversed – start with the lowest frequencies first, and finish the measurement with the highest frequencies. If you get the same impedance spectrum twice, it’s a good indication that the data is reliable.
Get involved
Three- and four-electrode measurements
959 words (5 min) Matthew Lacey 28 Mar 2023
Info
This content has been transferred from lacey.se and is not updated for this site yet.
In a previous page we looked at the Debye circuit as a model in polymer electrolyte characterisation. Keeping the cell components and cell design simple is a good strategy for getting simple, easily-to-analyse data. But what if you want to measure the impedance response of a complicated real system? Something like, a Li-ion battery, maybe, which has two electrodes essentially made of mixtures of electrochemically active powders?
In this case maybe the system is too complicated to be able to understand every process going on, especially when the electrochemical reactions are probably distributed very unevenly throughout the electrodes. But perhaps you just want to separate out the impedance of the positive electrode from the impedance of the negative electrode? This is useful in battery testing, because to be able to see the contributions of each electrode separately will give the tester some insight into, for example, whether one electrode is degrading faster than the other over long-term testing.
In principle, it is quite simple to do this by using a reference electrode, although practically designing and building a reliable cell might be more tricky. Here, I’ll share with you a demonstration experiment I did for some visiting students, as an example of separating out the contributions of the two electrodes in a small Li-ion test battery (since it worked almost perfectly on the first and only attempt!).
Essentially, I built a cell that looked like this on the inside:
It has a working electrode (positive electrode), which is a film of the battery active material mixed with carbon and a binder, and coated onto Al foil; a counter-electrode (negative electrode), which for simplicity in this case is just lithium metal foil; and two reference electrodes. Both reference electrodes are also made of lithium metal foil but cut into rings, which sit concentrically around the positive and negative electrodes – using references of this shape and placement means that the cell is as symmetric as possible, and gives as reliable impedance spectra as possible.
If you only want to measure the contribution from the positive and negative electrodes individually, you only need the one reference electrode. You’ll see why I’ve used two later on. There are also, of course, separators between the electrodes to prevent them from contacting each other inside the cell.
Why reference electrodes
If you’re a regular potentiostat user, you’ve probably noticed that it has four (or maybe more) leads with which you can connect up your electrochemical cells. Battery testing instruments too, usually have four leads, but usually connected up into pairs.
This is because the measurement of voltage and the transmission of current are divided between separate pairs of leads (also known as force and sense respectively). The current flows through the force leads, and the voltage is measured between the two sense leads. For battery testing (and usage), the force and sense leads for each electrode are simply connected to each other. This is a two-electrode measurement. If you separate the voltage measurement from one or both of the current-carrying electrodes, however, and measure the voltage between two different points (see the diagram on the right) – you can remove the contribution from those electrodes. This is the basis of the four-point probe.
If you are an electrochemist, you are probably familiar with the use of reference electrodes already. In that case, the aim is to place a stable voltage-sensing electrode as close as possible to the working electrode (to which the other sensing electrode is connected). The current flows through the counter electrode and the working electrode, but the potential drop across the cell is only measured between the working electrode and the reference electrode – the impedance associated with the counter electrode is not measured.
How does this work in practice?
Ok, let’s go back to the battery I made. I’ve drawn a simplified(!) equivalent circuit for the cell, and indicated where the reference electrodes are placed in relation to these different components.
Between the WE (positive) electrode and RE1 is the impedance of my battery electrode (with a reasonable equivalent circuit to describe it when it’s just been assembled – but I won’t go into why it looks like this, at least for now). Between RE2 and CE is the impedance of my lithium negative electrode, which can essentially be described by a parallel R-CPE unit which models the reaction:
I have to keep the force leads connected to the positive and negative electrodes of the cell of course, but if I move around the sense leads I can measure different contributions from the different components of the cell:
two-point measurement – I can keep the sense leads connected to the positive and negative electrodes along with the force leads, and leave the reference electrodes disconnected. This way, I’ll simply measure the impedance response of the entire cell.
three-point measurements – I can measure the potential between one of the cell electrodes and one of the references – if I measure between WE and RE1, I’ll just measure the impedance of the positive electrode. Alternatively, I can measure between CE and RE2, and measure the impedance of the lithium metal negative electrode.
four point measurement – or, I can go really crazy, and measure between RE1 and RE2. Then, the equivalent circuit is just a resistor, corresponding to the ionic resistance of the electrolyte between the two references.
Below are the Nyquist plots for these different combinations. Can you see how the different parts of the equivalent circuit combine?
Note: lithium metal is, unfortunately, not a very stable or well-behaved electrode, and shows some difficult-to-model behaviour towards lower frequencies.
Get involved
Fitting With Real Data
1366 words (7 min) Matthew Lacey 28 Mar 2023
Info
This content has been transferred from lacey.se and is not updated for this site yet.
I know from personal experience that knowing the basics well enough to interpret a very simple and well-defined experiment is one thing – but actually taking that knowledge and trying to make sense of the response of a complex ‘real’ system is another.
As equivalent circuits become more complicated, they become harder to fit correctly. Impedance software uses non-linear least squares regression to fit the data to the models, and they need reasonable values to start with to converge on a solution. The more complicated the model gets, the more likely the software is to fail in its attempt to fit the data – or it returns a fit which looks ok on the plot but has huge errors for some of the terms.
It’s this aspect of practical EIS which I think makes it hard to learn, and I don’t think there’s a lot of help out there for users who don’t have the luxury of being able to consult a local expert.
On the previous page, on three- and four-electrode measurements, I showed some real data from a Li-ion test battery just after assembly, and a rather complicated suggested equivalent circuit for it. You might have also recognised that the Nyquist plot for the positive electrode (taken from a 3-electrode measurement) was the same as I showed on the main page of this guide (shown again on the right) – and maybe you’re wondering where the equivalent circuit came from.
Well, the equivalent circuit I’ve chosen is somewhat empirical – and I’m not going to guarantee that it’s the most appropriate possible circuit – but I figured it makes a convenient example to show you how you can approach fitting a more complex dataset. On this page I’ll detail the process I used to analyse the data by eye and build up the model.
Equivalent circuit fitting in ZView
The screenshots I present here are from ZView, which can be downloaded from the software manufacturer, although the latest versions will only run in ‘demonstration mode’ – which is basically unusable – unless you have an expensive licence. Older versions (e.g. 3.2b) are ok, and Google may help you find it…
Ok, to the data. Let’s look at the spectrum above again, and pretend the fit line isn’t there. What are the obvious features? Well, there’s the slightly depressed semicircle at high frequency, and then the behaviour is pseudocapacitive at lower frequencies (lowest frequency is 100 mHz, incidentally).
Essentially, the strategy I use is quite simple: fit the obvious stuff first, and then build up the rest of the model around that. Have a look at the following screenshot (you can click on it to enlarge it):
ZView lets you use sliders to fit only a part of the data if you want, and this is invaluable. Also very useful is ZView’s “Instant Fit” tool, which will fit the selected data only to any of six very simple equivalent circuits, without needing any starting values from the user. So a good place to start here, especially if you have no idea what values might be appropriate, is to fit the semicircle with the Rs (CPE-Rp) circuit (bottom-right in the Instant Fit window). As you can see, the fit is pretty good, with small errors, so I’ll take the numbers from the Instant Fit and start building up my own model in “Equivalent Circuits”.
Now to fit the low-frequency tail. I could try to fit it with a constant phase element, but the phase is obviously changing, so it’s probably not appropriate. I can try to fit it anyway just to show that it doesn’t fit well, even though the errors come out to be rather small:
The advice I often give to people who ask me about EIS measurements is that it helps to know what the spectrum should look like, although that isn’t always possible. At this point I should probably make a guess based on what I’m actually measuring. Since the electrode is for the most part a Li-ion battery positive electrode, there’s Li ions in the material which are probably moving around in response to my applied potential. So I can try to put a finite space Warburg (or “open” Warburg, in ZView language) to model that finite diffusion behaviour, instead of that CPE I tried earlier.
Well, it’s not really much better, though it makes a bit more sense… and it does fit that very first bit of the Nyquist plot after the semi-circle a bit better (the very short 45° part where Z’ ~ 90 Ω), so maybe I’m onto something.
I also know that the electrode is porous, and that porosity can be represented by a finite length Warburg (or “short” Warburg, in ZView terms). So I can try to include this in the model, also in series with the FSW and the resistor R2. I fixed terms initially so that the software only guessed values for the -R (resistance) and -T (time constant) terms for the new FLW element (using 1 for each of the terms as a starting point), and then re-ran the fitting with other values freed up, keeping the phase of the Warburgs at 45° (P = 0.5).
So, now the fit is fairly decent – although the errors of 12% for the FSW element give me a little cause for concern. I could un-fix the phase angles as well – and I did, and the fit to the Nyquist plot looked better – but it caused some of the errors associated with the elements to hit more than 500%! In that case, you really can’t be sure the model is a good one.
Perhaps I can try something else. What if I try again with the Warburg elements in series with the CPE instead of in parallel with it?
Well, the fit in the Nyquist plot looks ok, similar to the last one, maybe except for that first bit of the tail… oh, wait. The values for the FSW (W1-T and W1-R) have become absurdly small and the errors are 50 million percent! That’s a telltale sign of having screwed up.
Wrap-up
Well, I concede I probably haven’t reached anything conclusive here, but if you’re new to EIS you now hopefully have at least a bit of an idea of how you can approach an equivalent circuit fitting, and some appreciation for some of the difficulties.
It’s an anticlimax, I suppose, but I do have a reason. When I was a lab teacher for EIS at the Southampton Electrochemistry Summer School, we ran a few very well-designed experiments which we knew would give easy-to-understand results; the most in-depth experiment was an analysis of the ferricyanide redox couple at a mirror-smooth glassy carbon electrode, where you get a very lovely Randles circuit response and it’s dead easy to fit and extract a load of meaningful parameters from. This is great for learning about the technique, but of course the delegates – mostly not academics – ultimately wanted to know how to apply it practically to their own work. We often got questions about predicting or analysing impedance spectra of much more complex systems like these, and we weren’t always able to give a clear answer.
The truth is that real-world EIS is more like this example here, in my experience. The more you want to learn the more you have to think about the design of the experiment, sometimes you have to take an educated guess at an appropriate equivalent circuit, and even then it can be difficult to be sure. Hence this example – it was genuinely my first attempt at fitting this data, so this was my real thought process.
Perhaps I’ve ended this example pessimistically. I don’t mean to – EIS is still a very useful technique. It’s fast, you get a lot of information that few other techniques can give you as easily – but sometimes you’ve got to be realistic about how much quantitative analysis you can do with it, and careful that when you do that analysis, the model and numbers make sense.
Get involved
Recommended Reading
623 words (3 min) Matthew Lacey 28 Mar 2023
Info
This content has been transferred from lacey.se and is not updated for this site yet.
The following are a selection of papers which I think are especially useful for further reading on specific topics. For the most part they are quite focused, and elaborate on some of the topics discussed in the pages on this website. The list is by no means exhaustive, and if you have any suggested papers which you think could find a place in this list, please feel free to suggest them to me either by email or in the comments below.
R. de Levie, “On porous electrodes in electrolyte solutions” - key fundamental work describing the original transmission line model for porous electrodes, diffusion within pores and charge transfer reactions within porous electrodes. (with thanks to Jeremy Meyers for the suggestion)
If you’ve studied all of the previous pages in this series, why not put your new-found skills to the test with a little exercise?
Below are three plots of simulated impedance data. The data is representative of a symmetrical electrochemical cell, comprising a thin lithium-conducting polymer electrolyte with relatively poor ionic conductivity, sandwiched between two lithium metal electrodes. (Hint: this sort of system is discussed here and here.)
Exercise: from the plots below, can you propose an equivalent circuit? Once you have come up with a circuit model, can you calculate all the relevant parameters… by hand?
This is a tricky exercise. See how far you get - and if you’re done or can go no further, why not try your hand at fitting the “raw” data? You can download the data here.
A little about this exercise
This exercise is a variant of one I used in teaching on a number of occasions. There are several aims here, which I believe test some of the most important skills for building real competence in the technique:
To understand how to propose a reasonable equivalent circuit, based on what one knows about the system under study and what features can be seen in the data
To understand how impedance data can be plotted and what information it provides
To be able to calculate parameters by hand, at least as a way to give initial guesses to the fitting software, so it can refine the fit for more complex models
Want to check your answers? You can check how you did against the real circuit and parameters by following this link. Don’t cheat!
Get involved
Transference number measurement
1701 words (8 min) Matthew Lacey 28 Mar 2023
Info
This content has been transferred from lacey.se and is not updated for this site yet.
The most common experimental method for measuring t+ in a polymer electrolyte is the so-called Bruce-Vincent method, named for the work done by Colin Vincent and Peter Bruce on this subject, published in 1987 (here and here).
The method involves the polarisation of a symmetrical cell (i.e., an electrochemical cell with two Li metal electrodes) by a small potential difference, to induce a small concentration gradient, until the system reaches a steady state, with a concentration gradient that does not change further with time:
This method assumes - as I previously mentioned - that the ions in the electrolyte are perfectly dissociated. More specifically, that the electrolyte obeys the Nernst-Einstein equation, which relates the conductivity (and the electrical mobility) of an ion to its diffusion coefficient:
The derivation of the following equations is long and rather complex (the paper detailing the derivation is 17 pages long), but I will summarise here briefly.
In this case, assuming a perfect system, the current that flows initially depends only on the conductance of the cell and the potential difference
$\Delta V$:
$$I_0 = \frac{\sigma}{k} \Delta V$$
where
$k$ is the cell constant, i.e., the ratio of the distance between the electrodes to the surface area. At steady state, the concentration gradient does not change with time. The migration of the anion is exactly balanced by its diffusion in the opposite direction (that is, the net flux of anions is zero - this has to be the case, because the electrode surfaces are “blocking” to the anions). Meanwhile, the current carried by Li+ is carried exactly 50:50 by migration and diffusion.
The end result of all this is that for very small potential differences (< 10 mV), the current flow at steady state is given simply by:
$$I_{ss} = \frac{t_+ \sigma}{k} \Delta V$$
Or, indeed:
$$t_+ = \frac{I_{ss}}{I_0}$$
However! In real systems we have interfacial resistances resulting from surface layers and charge transfer kinetics, so we cannot apply this directly. These resistances may also change with time and concentration. We need to measure the interfacial resistance both initially and at steady state, and ideally the impedance spectra will look something like this:
In this case the series resistance Rs is the ionic resistance of the electrolyte, and Rp the interfacial resistance. Now, the initial current is given by:
$$I_0 = \frac{\Delta V}{k/\sigma + R_{p,0}}$$
Correspondingly, the current at steady state is given by:
$$t_+ = \frac{I_{ss}\left(\Delta V - I_0 R_{p,0}\right)}{I_0 \left(\Delta V - I_{ss} R_{p,ss}\right)}$$
This is the well-known Bruce-Vincent equation. Simple, right?!
Determining transference numbers
Because of the assumptions, the Bruce-Vincent method is only valid in very (and unrealistically) dilute systems. So how can we determine the real transference number?
One of the established and reliable methods for calculating the true transference numbers is the Hittorf method. In this case, a known amount of charge is passed through a cell, and the electrolyte is then divided into four or more sections (this is a bit easier with a polymer electrolyte!).
The reason it needs to be four or more is that there must be at least two reference sections where the concentration of the salt are identical to each other. During the passage of charge, migration transfers cations and anions to the electrode surface. If the passage of charge is stopped before a concentration gradient forms in the reference sections, then the transfer of cations and ions into the other sections is due only to migration, and not diffusion. No assumptions about the nature of the electrolyte need to be made!
In this case, there is a change in the amount of salt in section 1 in the diagram above according to:
And therefore, if you can determine the concentration of salt in the sections, you can calculate T- and then, accordingly, T+.
Bruce, Hardgrave and Vincent did in fact use this method in 1992 to show that for a PEO polymer electrolyte containing LiClO4 salt, T+ was calculated to be 0.06 ± 0.05 by the Hittorf method, compared with ~0.2 by their own Bruce-Vincent method. This result showed clearly how the transport of neutral ion pairs and/or negatively charged triplets can cause an overestimate of the true transference number in the Bruce-Vincent method.
There are conceptually similar methods currently under development based on electrophoretic NMR, which also allow for determination of the true transference number, but these approaches are not yet accessible for most researchers. Also, this method (at the time of writing) has yet to be applied to solid polymer electrolytes.
John Newman and co-workers have also been developing an electrochemical method based on concentrated solution theory which also does not require any prior assumptions about the electrolyte. They have shown that under the same DC polarisation conditions as in the Bruce-Vincent experiment, that for a concentrated electrolyte:
$$\frac{I_{ss}}{I_{0}} = \frac{1}{1 + Ne}$$
where
$Ne$, the dimensionless “Newman number” is a more complicated term containing the true transference number:
To use this method, several things need to be measured:
$\frac{I_0}{I_{ss}}$, as in the Bruce-Vincent method; the ionic conductivity,
$\sigma$; the salt diffusion coefficient,
$D$, which can be obtained from the voltage of the cell as it relaxes after the DC polarisation; and the “thermodynamic factor”,
$\left(1 + \frac{d \ln \gamma_\pm}{d \ln m}\right)$, which quantifies the concentration dependence of the activity of the ions, and can be obtained from measurements on a concentration cell.
This is a lot of things to measure, and there are different errors associated with all of the measurements; so the overall determination of T+ is subject to significant experimental error. Nonetheless, Pesko et al. have recently used this approach to demonstrate that T+ becomes negative in highly concentrated PEO:LiTFSI polymer electrolytes.
What does this mean for the Bruce-Vincent method?
The Bruce-Vincent method is so convenient, and has become so widely used in the polymer electrolyte research area, that it has effectively become a standard technique used in most reports on new materials. Because of this, there are probably a lot of researchers - maybe even most - using it without fully understanding it.
This is especially relevant now, where a lot of research effort is directed towards, for example, electrolytes with very high salt concentration - where we are a long way from ideality and transference numbers are very high (or so it would appear)! This is also relevant in systems based on ionic liquids, where there are only ions in the electrolyte, and Li+ transference has to compete with the transference of other positively charged species.
In these cases, clearly the Bruce-Vincent method does not give the transference number, T+, or indeed the transport number, t+ - not least for the fact that it measures processes occurring which are not either of those things.
Bruce and Vincent themselves recognised this was the case very shortly after publishing their method, and suggested in such cases that the result of their method should instead be termed the “limiting current fraction”, F+. I would tend to agree - it surely doesn’t make sense to report some quantity you have measured as being T+ when you know it can’t be.
Is F+ a useful quantity, and what does it mean? Well, this debate is likely to continue long into the future. For what it’s worth, I think it can be a helpful value to allow comparisons between different materials (you would expect in most cases that a higher F+ would be a good thing), and to compare results between different groups.
But, F+, as we saw in the expression for the Newman number before, is a mash-up of several other different properties, and so therefore it doesn’t mean very much in itself. For modelling purposes, it’s hopeless - the real T+ is needed.
The problem is, it’s not easy to get. As appealing as the assumption-free Newman approach looks, I’m cautious: it’s not that much more work, but the errors can get pretty large, the T+ values don’t always seem to match up with the Bruce-Vincent method in the very dilute case where you expect they should agree, and I would like to see the results backed up by other methods such as the Hittorf cell or electrophoretic NMR to demonstrate its robustness.
I reckon the Bruce-Vincent method is here to stay, but its users could probably do the field a favour by not calling the result of the experiment something that it isn’t.
Wrap-up
Hopefully this article has been helpful in making sense of the minefield of transference and transport in battery electrolytes. If I would pick out any specific points as particularly important, I would say:
There is a difference between transport, t, and transference, T. And they are often mixed up, so it’s a good idea to know how to spot this. Electrochemistry textbooks like Bard & Faulkner discuss transport, but not transference; polymer electrolyte research papers often discuss transference, not transport, but both use small t to denote this. It’s confusing, and I suggest using T for transference to be clear about this!
Know what the assumptions in the Bruce-Vincent method are - no ion association (Nernst-Einstein equation is valid), and only applies to very small DC polarisations (< 10 mV).
If you know that you have a system which behaves non-ideally (that would be essentially all of them), then consider using F+ instead of T+ to report the results of a Bruce-Vincent experiment.
Whoever invents a simple, convenient, accurate method for measuring true transference numbers of both solid and liquid electrolytes is going to be a very well cited scientist indeed.
Get involved
MacMullin number measurement
806 words (4 min) Marcel Drüschler 05 Aug 2023
The MacMullin number is an important quantity which describes the effect of a battery separator on the electrolyte solution in which it hosts.
The MacMullin number, NM, tells you the factor by which the conductivity of an electrolyte-wetted separator foil is reduced compared to the conductivity of the “free” electrolyte solution. That is, if you have a MacMullin number of 10 and the bulk, or “free”, electrolyte solution has a conductivity of 10 mS/cm, the effective conductivity of the electrolyte-wetted separator is 1 mS/cm, or, reduced by a factor of 10 compared to the bulk electrolyte
The MacMullin number is influenced by three factors: the porosity, the tortuosity (which depends on pore size, pore size distribution, and inter-pore-connectivity), and the thermodynamics of the electrolyte-separator material interface. Depending on the selected physical model, there are various formulae you can find in the scientific literature linking the MacMullin number in particular to the porosity and tortuosity[1]. Typically, most of these expressions look like the following generalized relationship:
$$N_M = \frac{\tau^n}{\varepsilon^m}$$
where
$\tau$ is the tortuosity and
$\varepsilon$ is the porosity of the separator.
Experimental procedure
To experimentally determine the MacMullin number, electrochemical impedance spectroscopy (EIS) can be used in combination with an elegant, simple, and robust experimental approach: the so-called stacking method: The single steps of the method are as follows[2,3]:
Punch out multiple specimens of your separator material, at least 4 pieces.
Store the speciments in electrolyte solutions. Depending on the wettability, only a few minutes might be needed while in other cases several hours are required.
Put the wetted separator foil between two current collector electrodes, e.g. made of stainless steel 1.4404.
Record an impedance spectrum (potentiostatic mode; 1 MHz-1 kHz is usually sufficient, with an ac-voltage amplitude of 5-10 mV (rms)).
Put another wetted separator foil on top of the other one.
Record another impedance spectrum for the two specimens being placed on each other.
Repeat that procedure at least four times.
Data analysis
You now have to evaluate the impedance data, which is typically done by equivalent circuit fitting. For an electrolyte-wetted separator foil, a model circuit consisting of a serial connection of an inductor I (cable contribution), a resistor R (ion migration through the electrolyte volume, but also contributions by cable and contact resistances), and a constant phase element CPE (= non-ideal capacitor; formation of an electrochemical double layer at the electrolyte-electrode interface) is often applicable.
Plotting the R-values against the number of separator layers results in a straight line with the slope being identical with the resistance for ion transport per separator layer Rion.
Knowing the thickness dseparator of the material and the contact area Acontact between the separator material and the current collector electrode, the conductivity of the electrolyte-wetted separator material can now be calculated:
The result is the MacMullin number. Typcial values for Li-ion battery separator materials are 4 – 20.
Please note that the MacMullin number is only a “constant” for a fixed separator-electrolyte combination. Once the electrolyte is altered, the MacMullin number can also vary[3]. The reason is that the MacMullin number is not only a function of the materials porosity and tortuosity, but also of the thermodynamics of the electrolyte-separator material interface; that is, the surface energies.
Relationship to other metrics
One might now ask the question: Why do you typically not find the MacMullin number on the specification sheet of separator materials?
You will find instead the Gurley number. The determination of the Gurley number is much simpler, faster, and cheaper, although the Gurley number only tells you something about the gas permeability of your separator material which is also related to the porosity and tortuosity, but which does not necessarily show a 1:1 correlation with the MacMullin number.
The determination of the MacMullin number requires working with chemicals, and therefore needs trained persons and a well-equipped chemical laboratory.
The determination of the MacMullin number requires relatively expensive measurement devices and test cells.
The MacMullin number is a function of the selected electrolyte and there is no generally accepted reference system. You would have to repeat the measurement with several electrolyte systems.
Supporting literature
[1] M. J. Martínez, S. Shimpalee, J. W. Van Zee, Measurement of MacMullin Numbers for PEMFC Gas-Diffusion Media, J. Electrochem. Soc., 2009, 156 (1), B80-B85.
[2] R. Raccichini, L. Furness, J. W. Dibden, J. R. Owen, N. García-Araez, Impedance Characterization of the Transport Properties of Electrolytes Contained within Porous Electrodes and Separators Useful for Li-S Batteries, J. Electrochem. Soc., 2018, 165 (11), A2741-A2749.
[3] J. Landesfeind, J. Hattendorff, A. Ehrl, W. A. Wall, H. A. Gasteiger, Tortuosity Determination of Battery Electrodes and Separators by Impedance Spectroscopy, J. Electrochem. Soc., 2016, 163 (7), A1373-A1387.

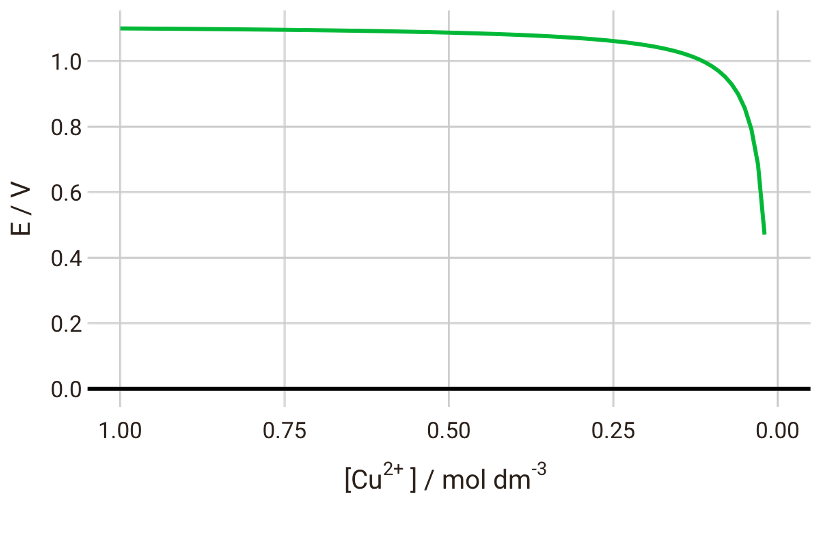



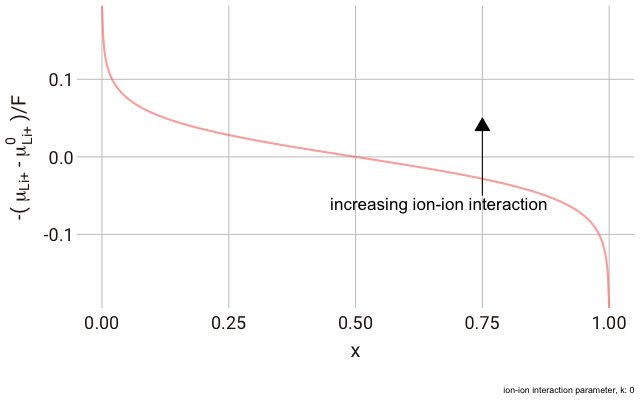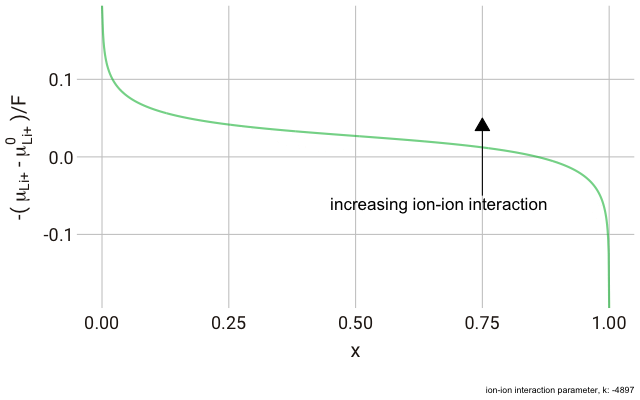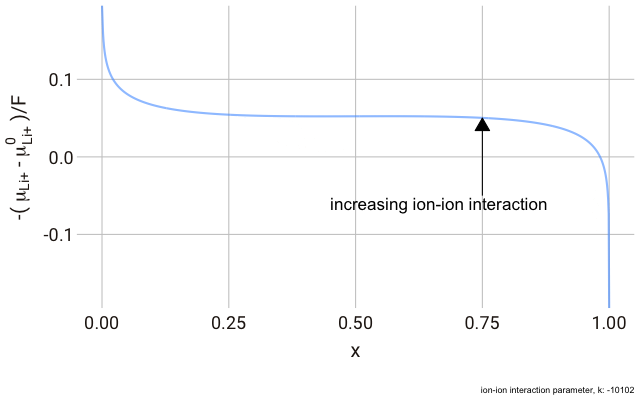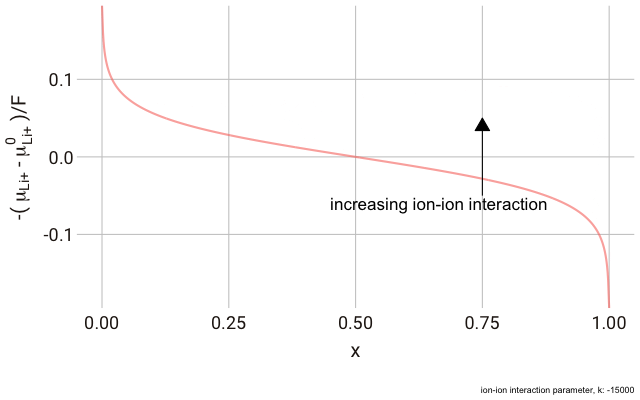
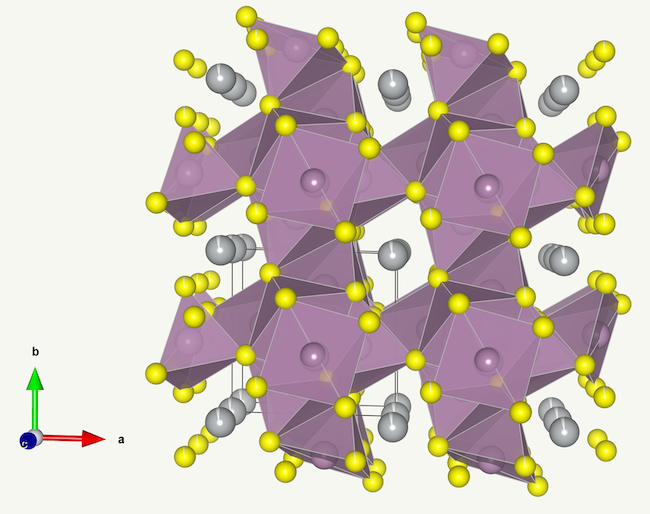
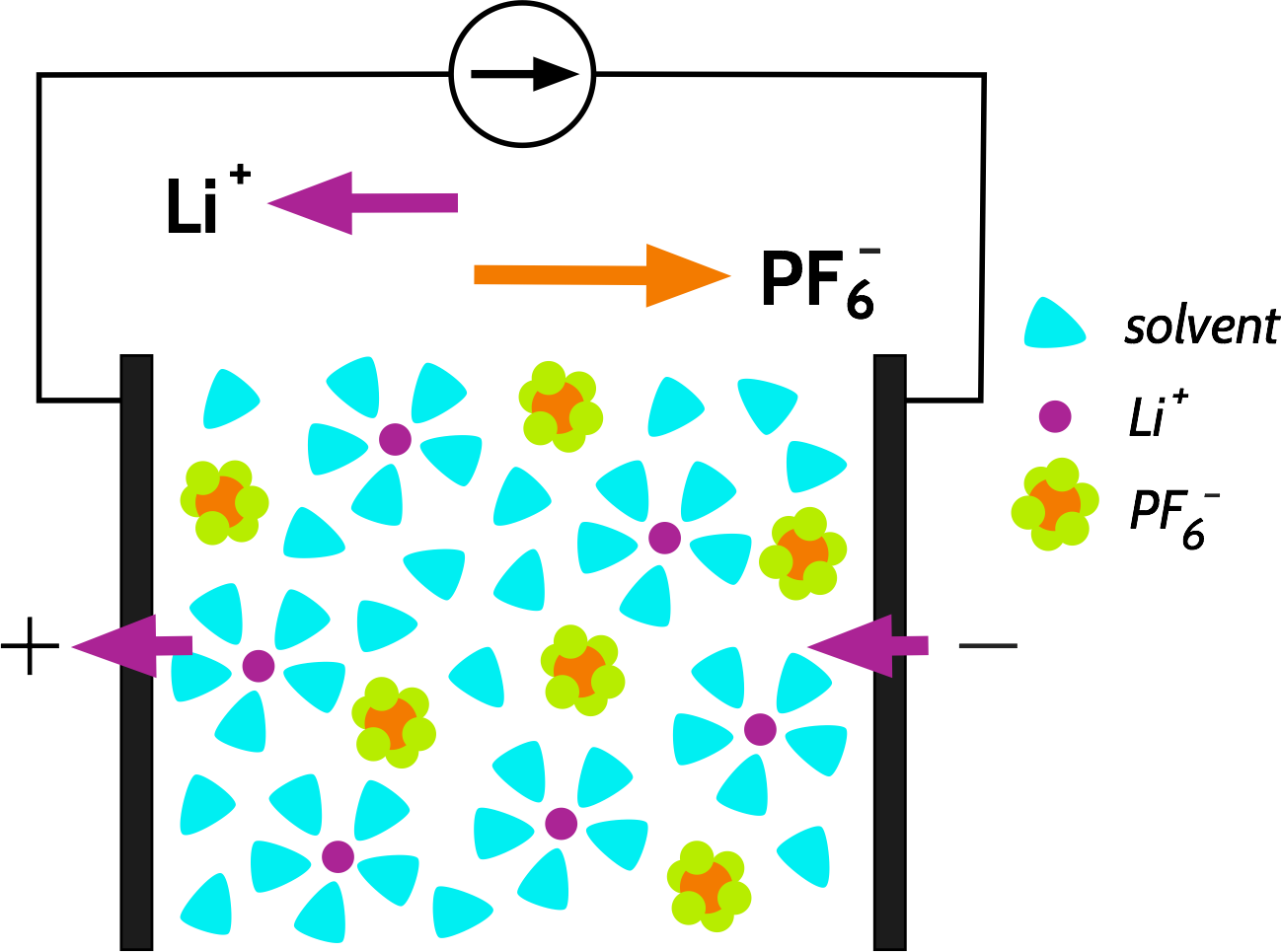

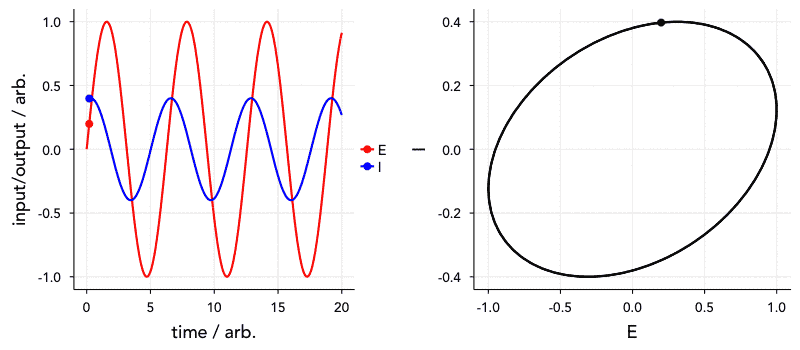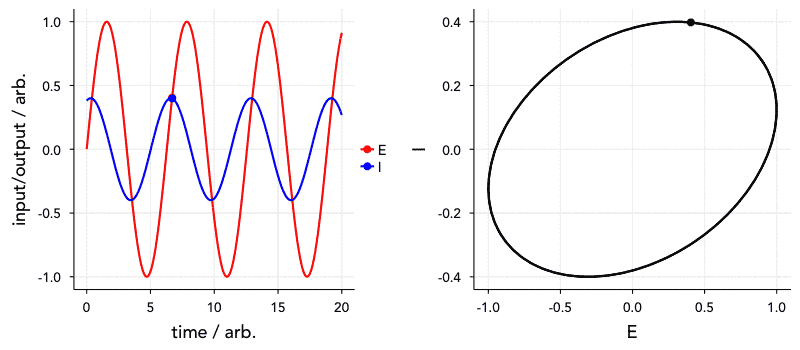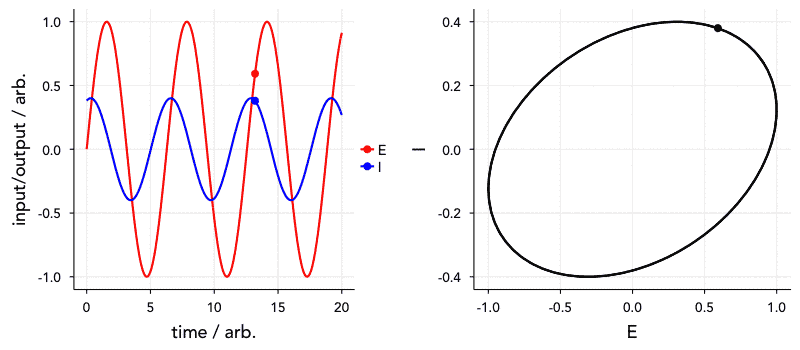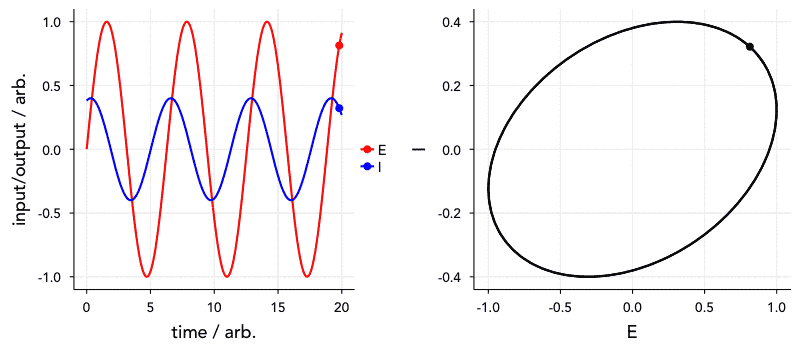












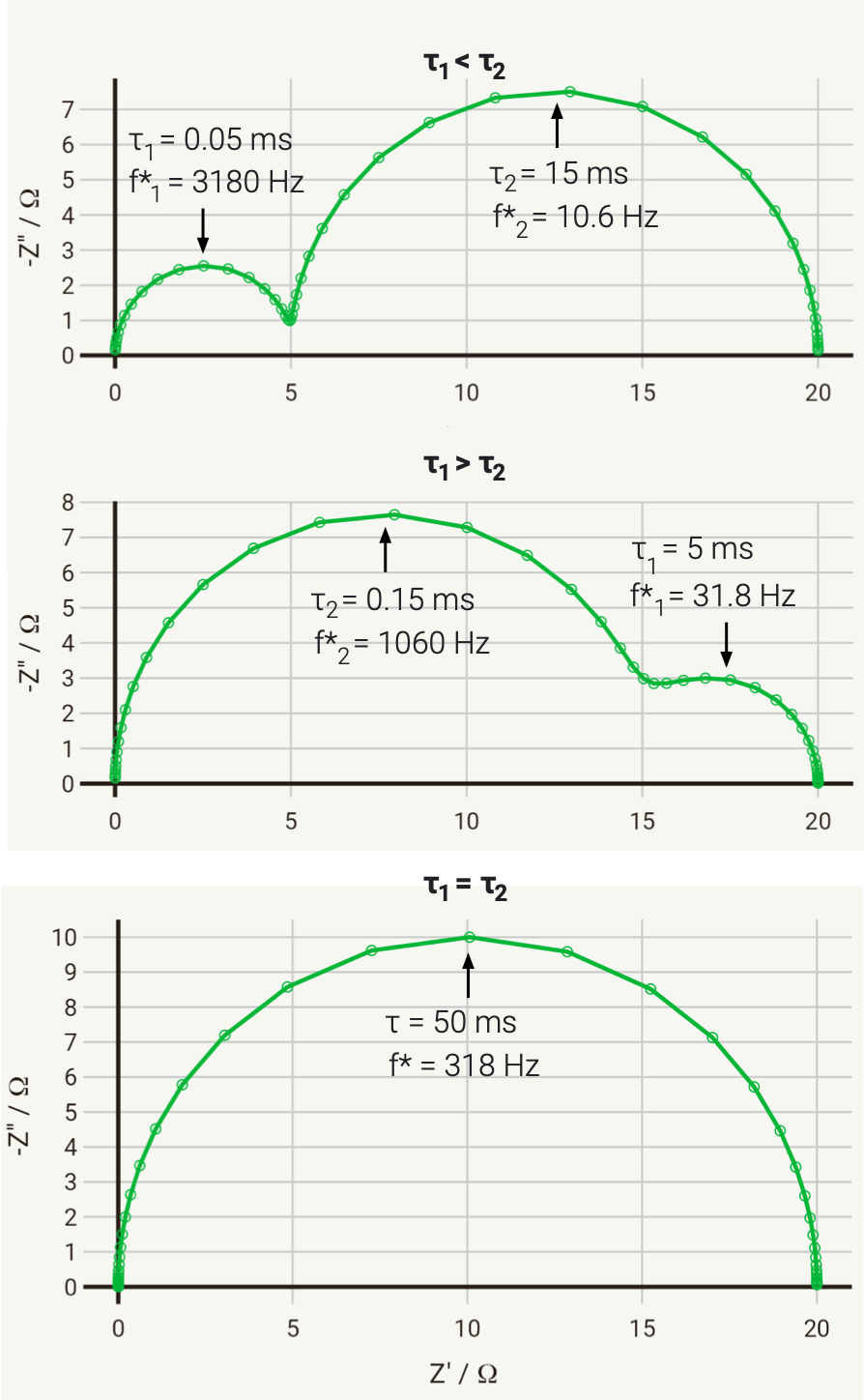
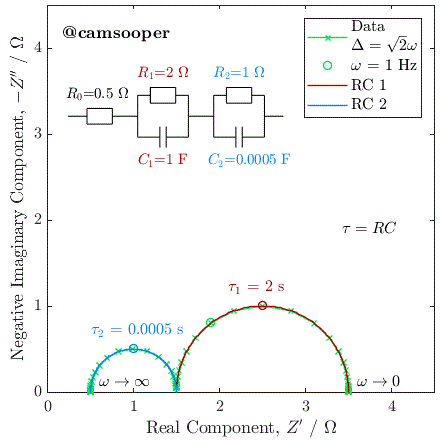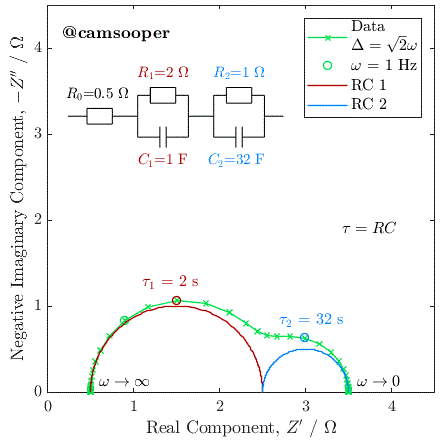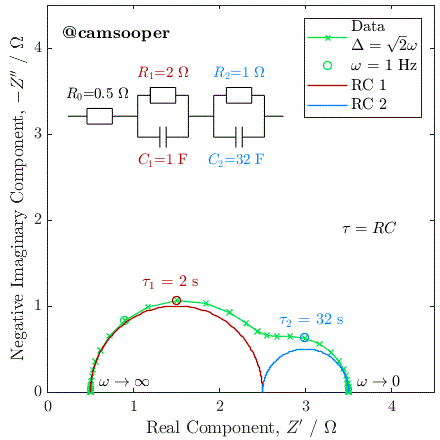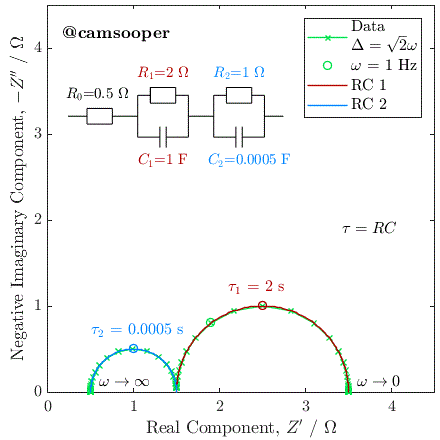











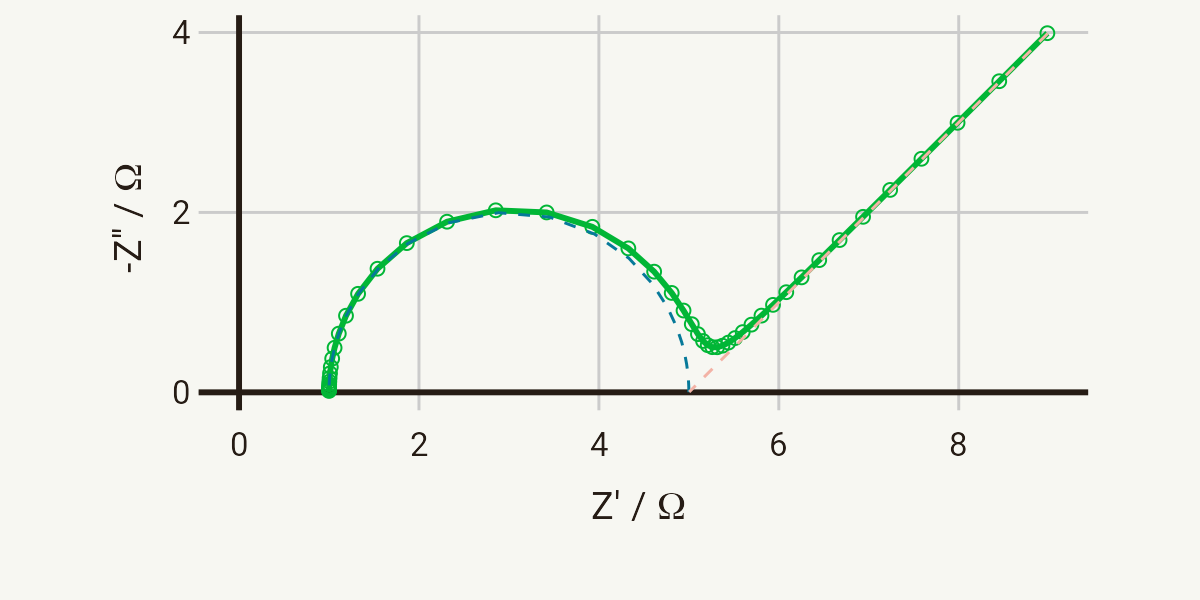
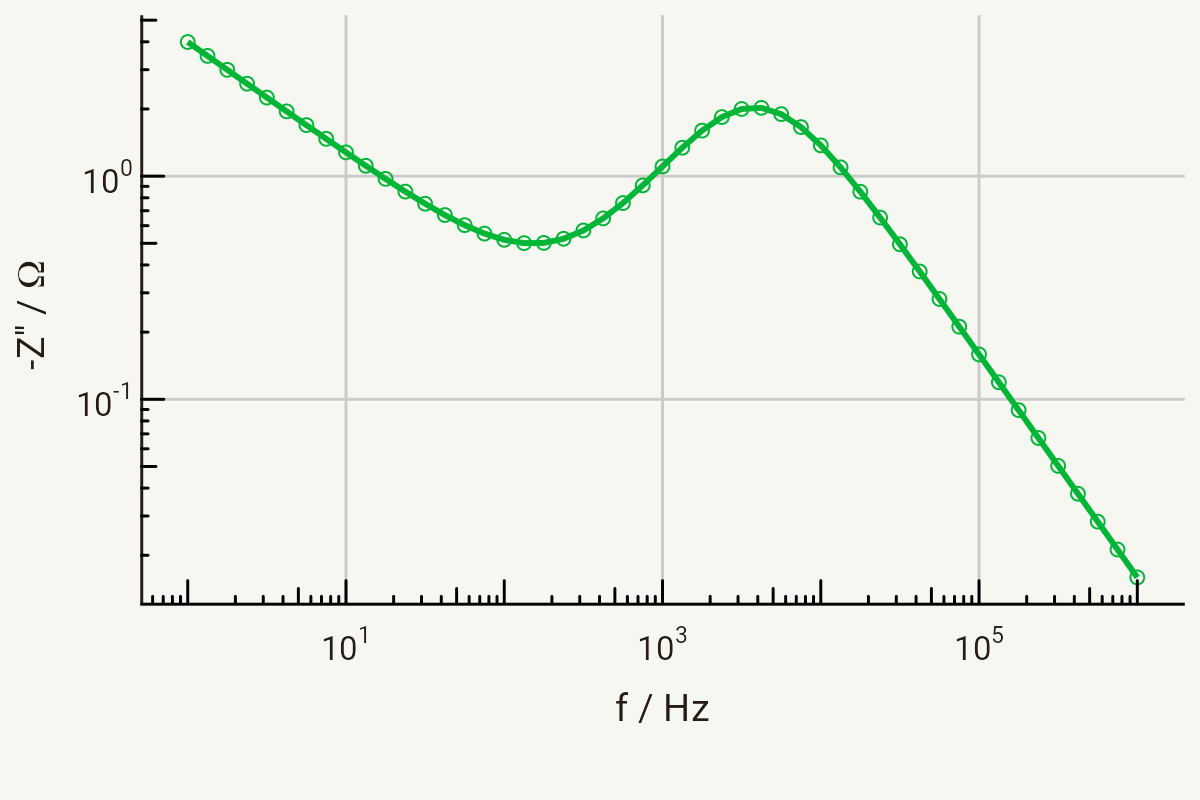

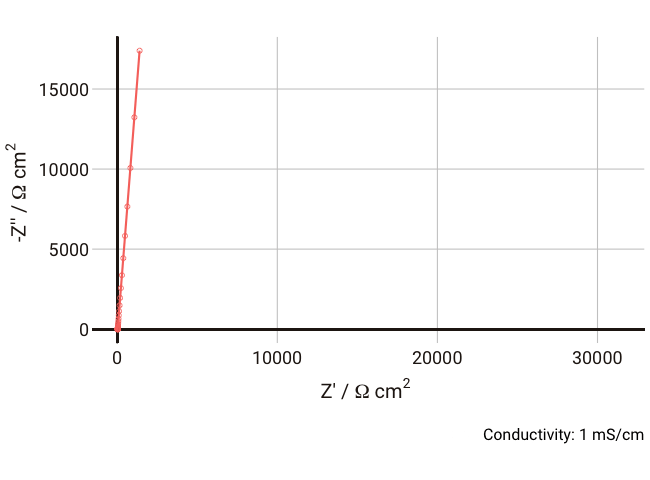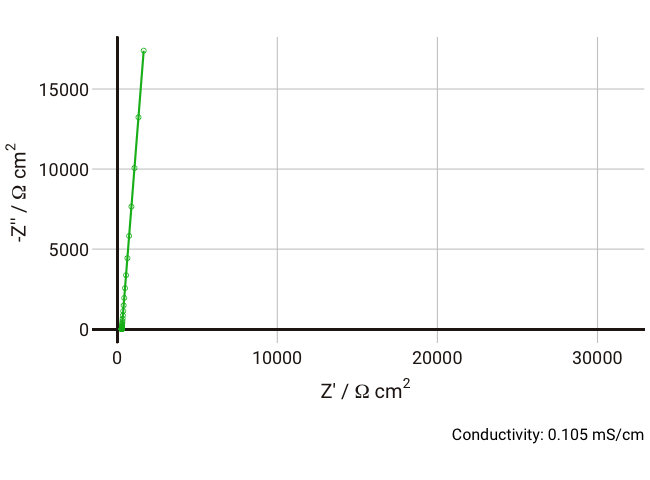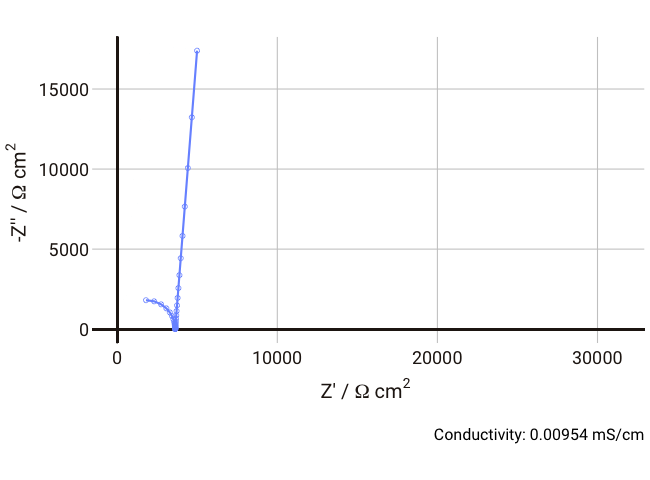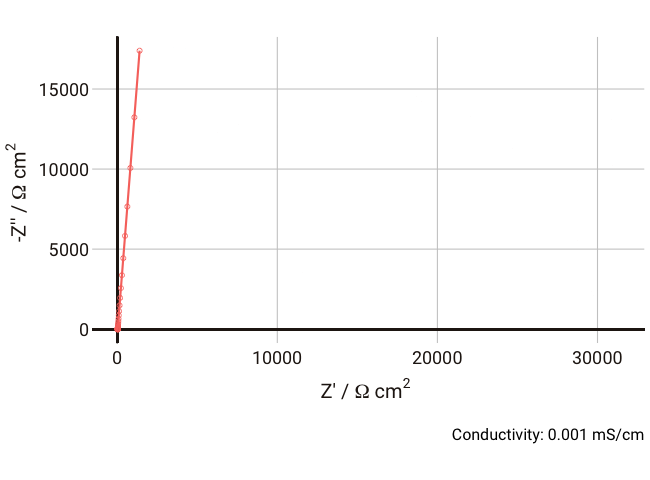



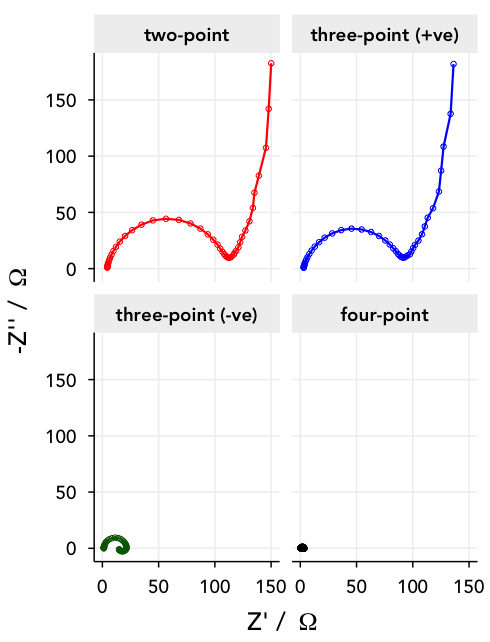
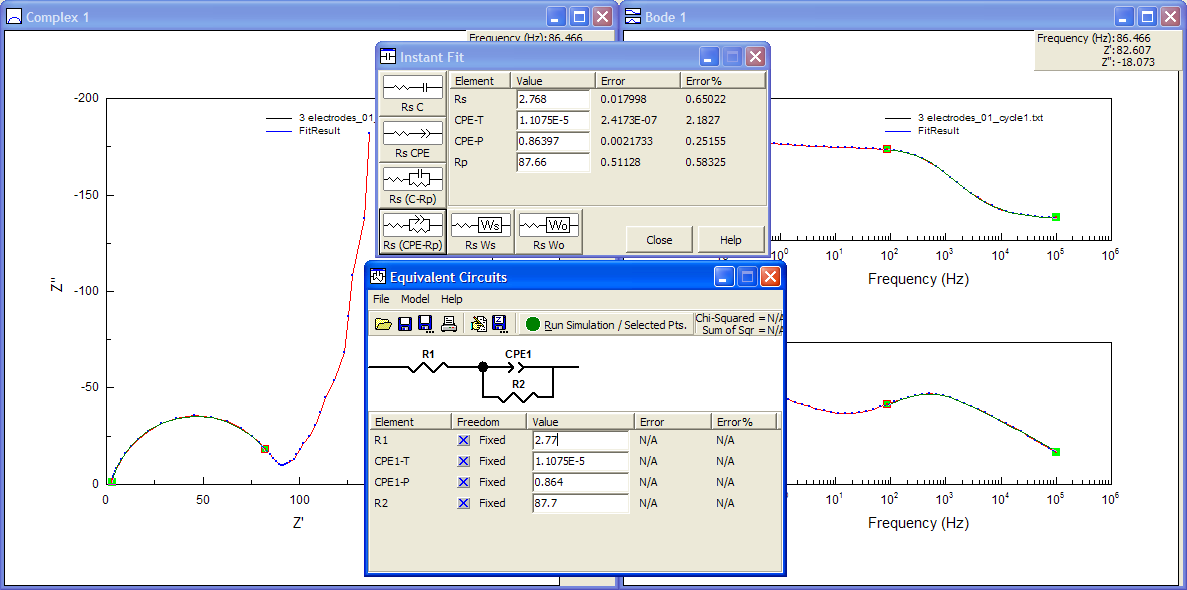

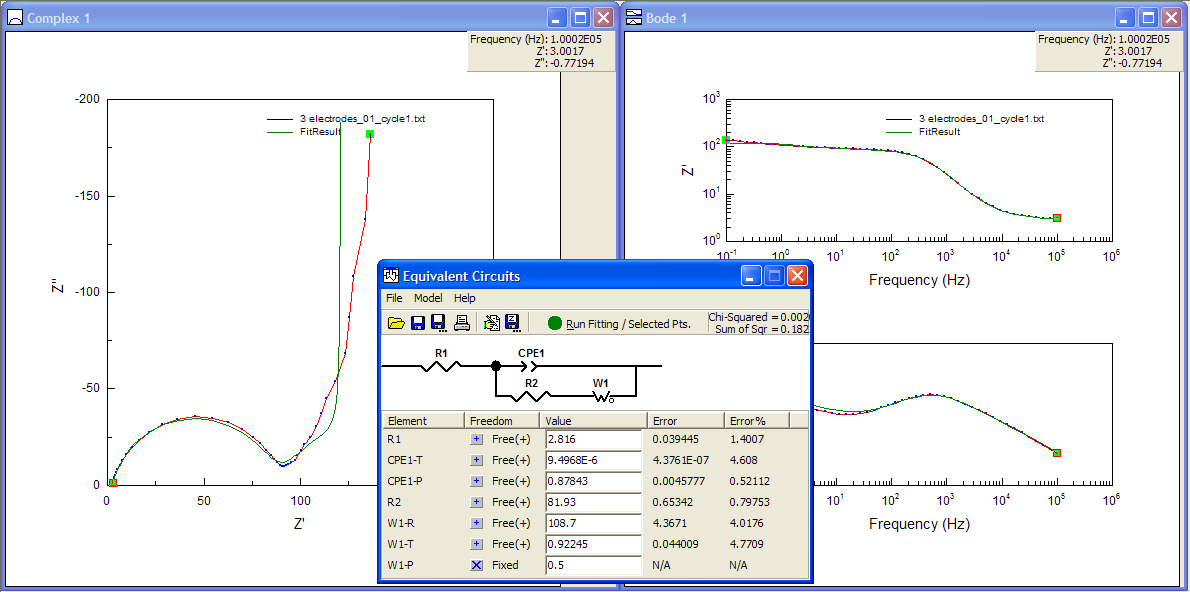
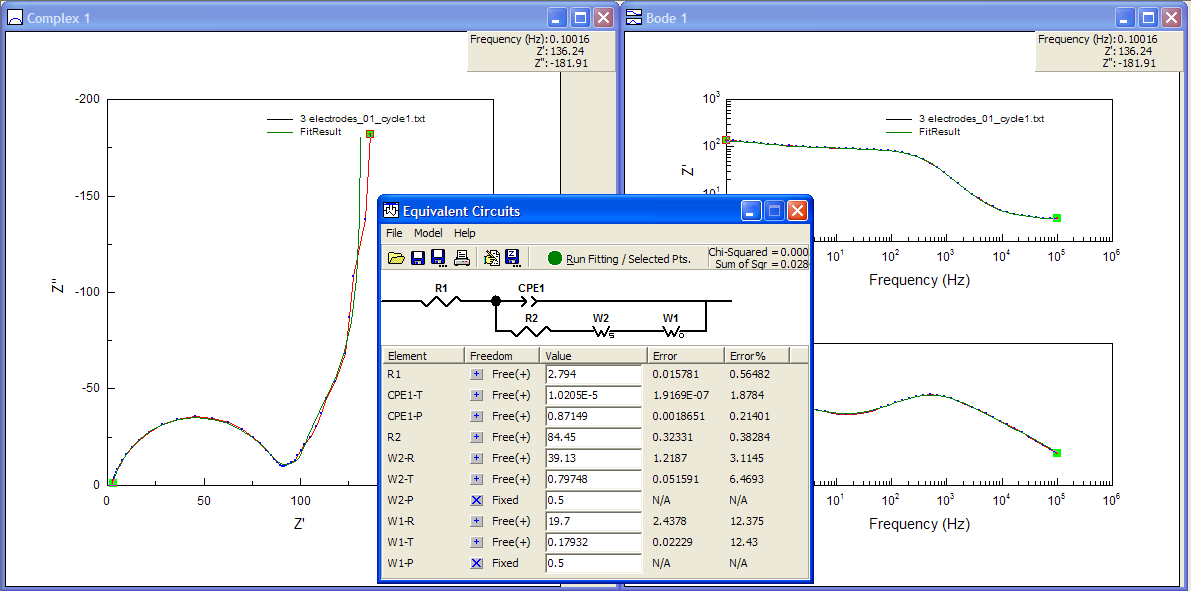
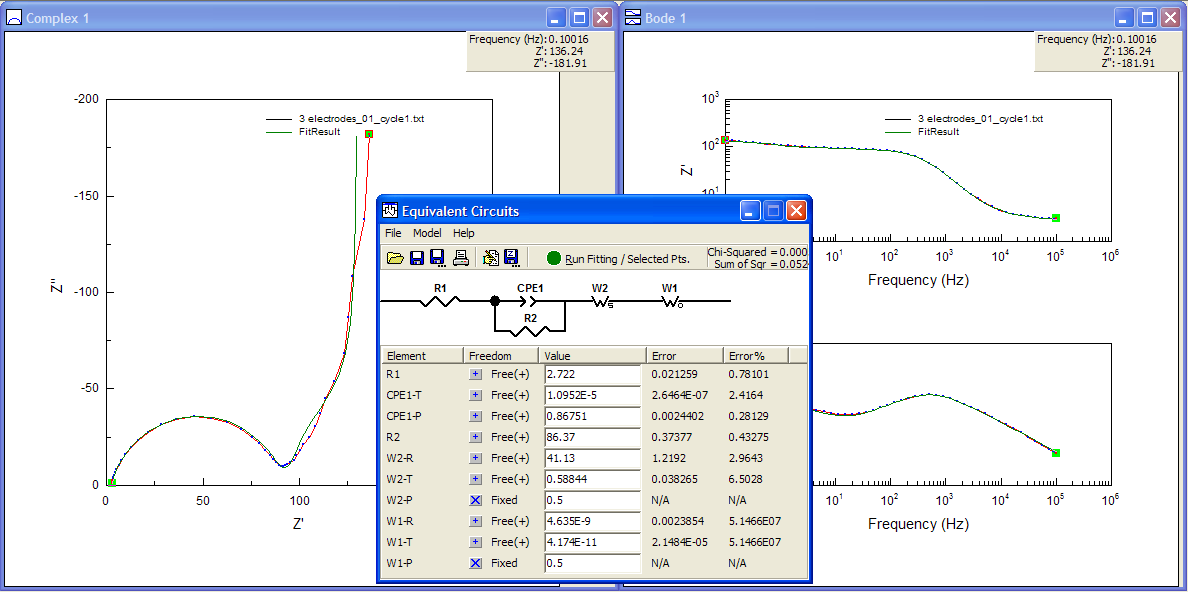
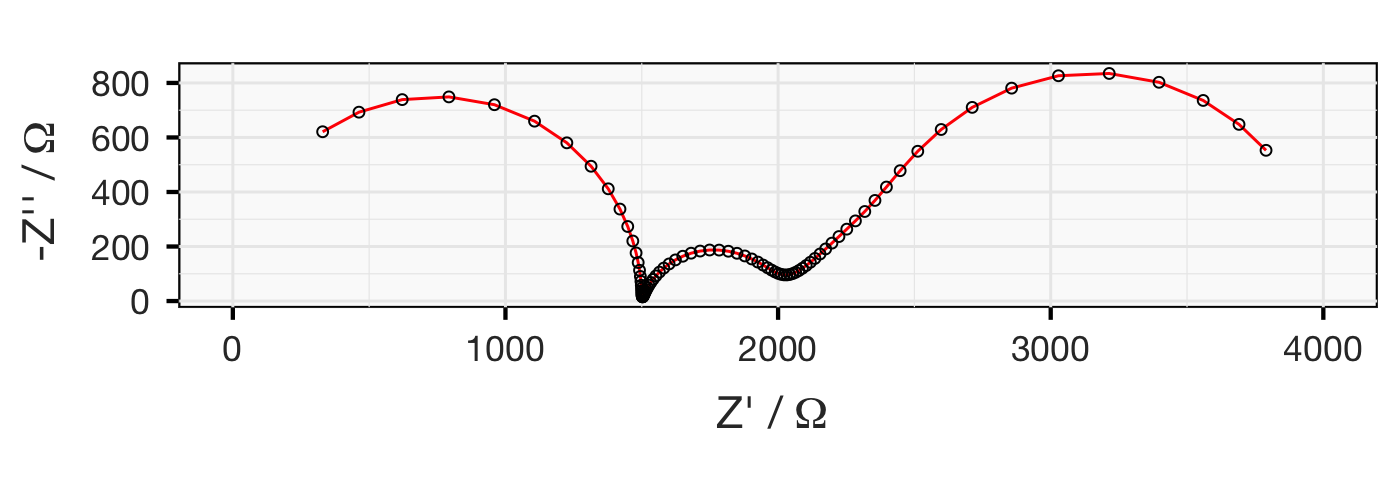
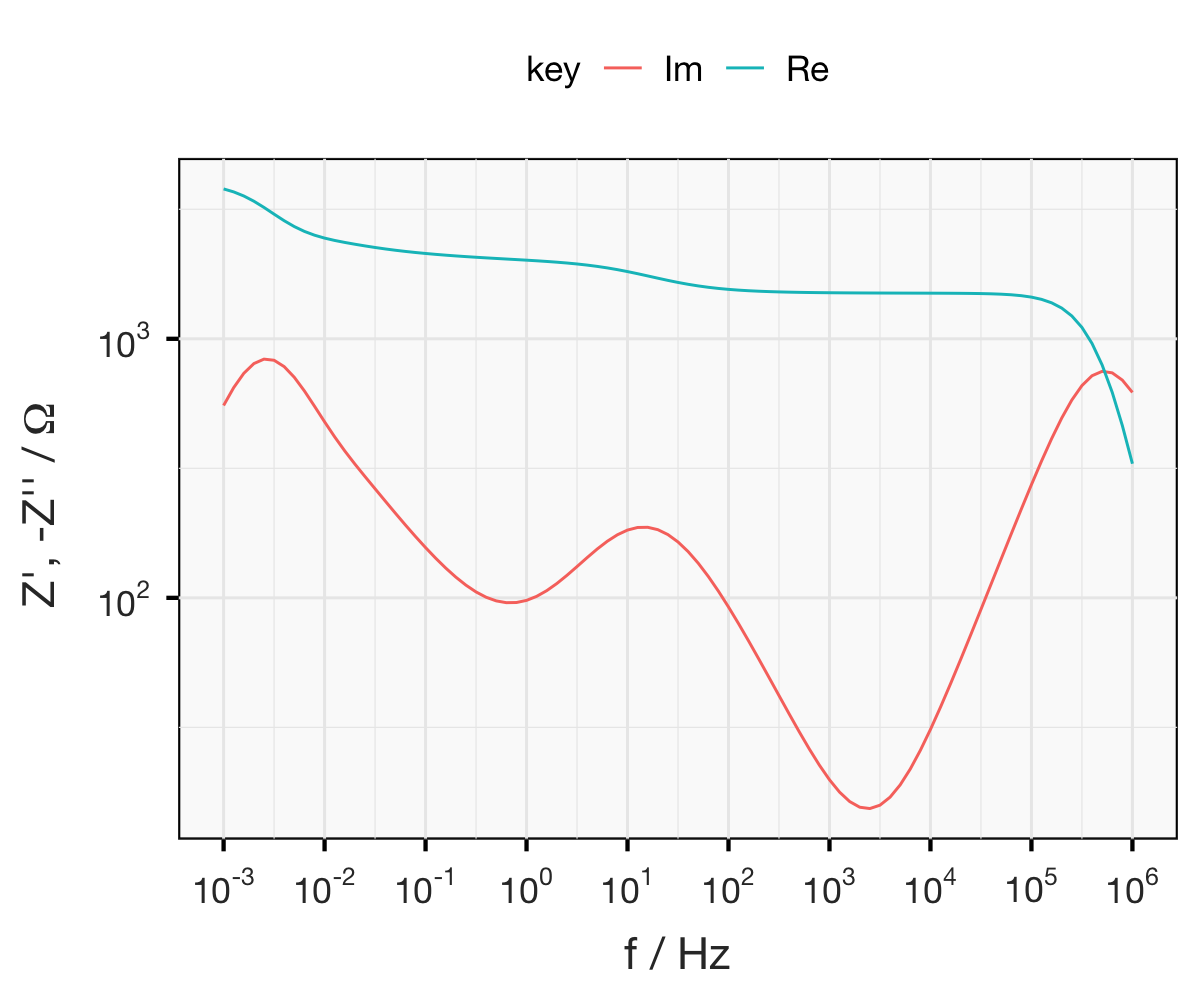
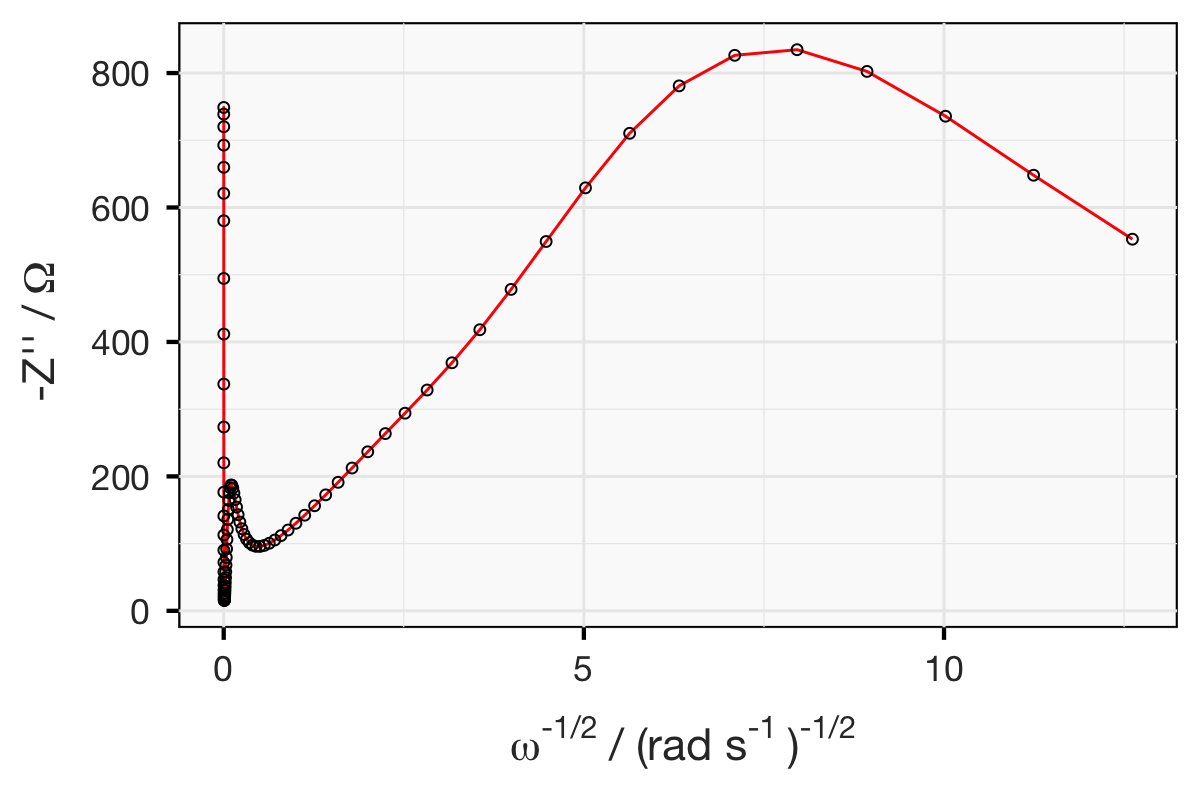



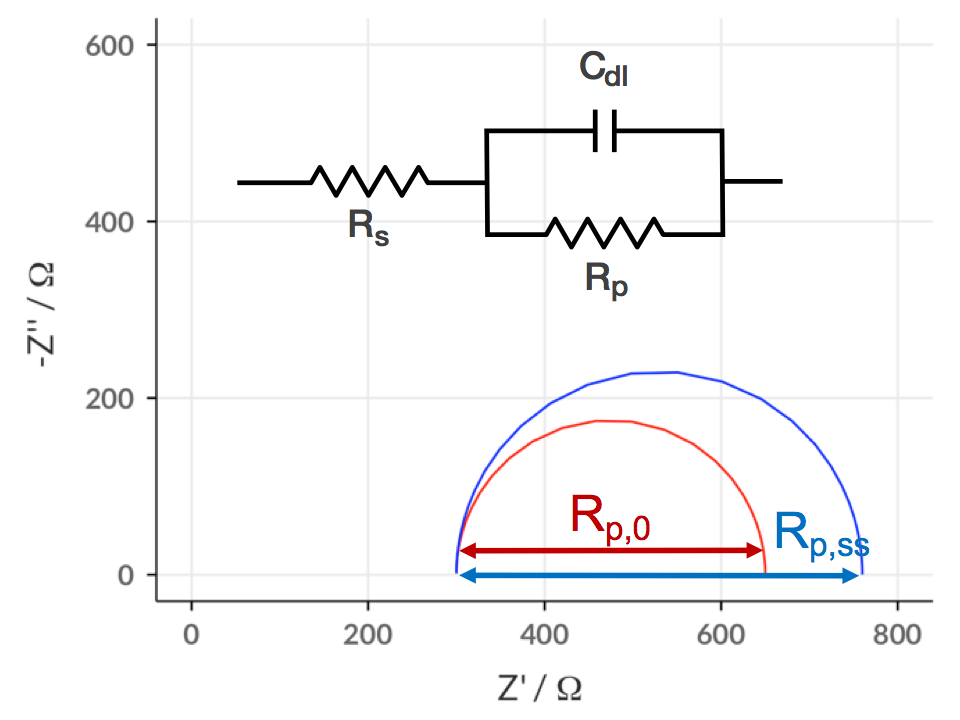

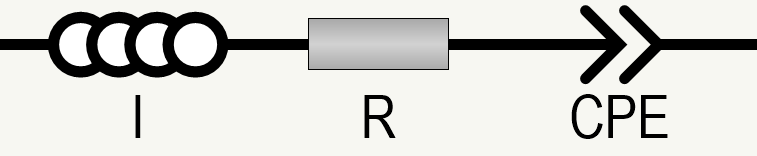

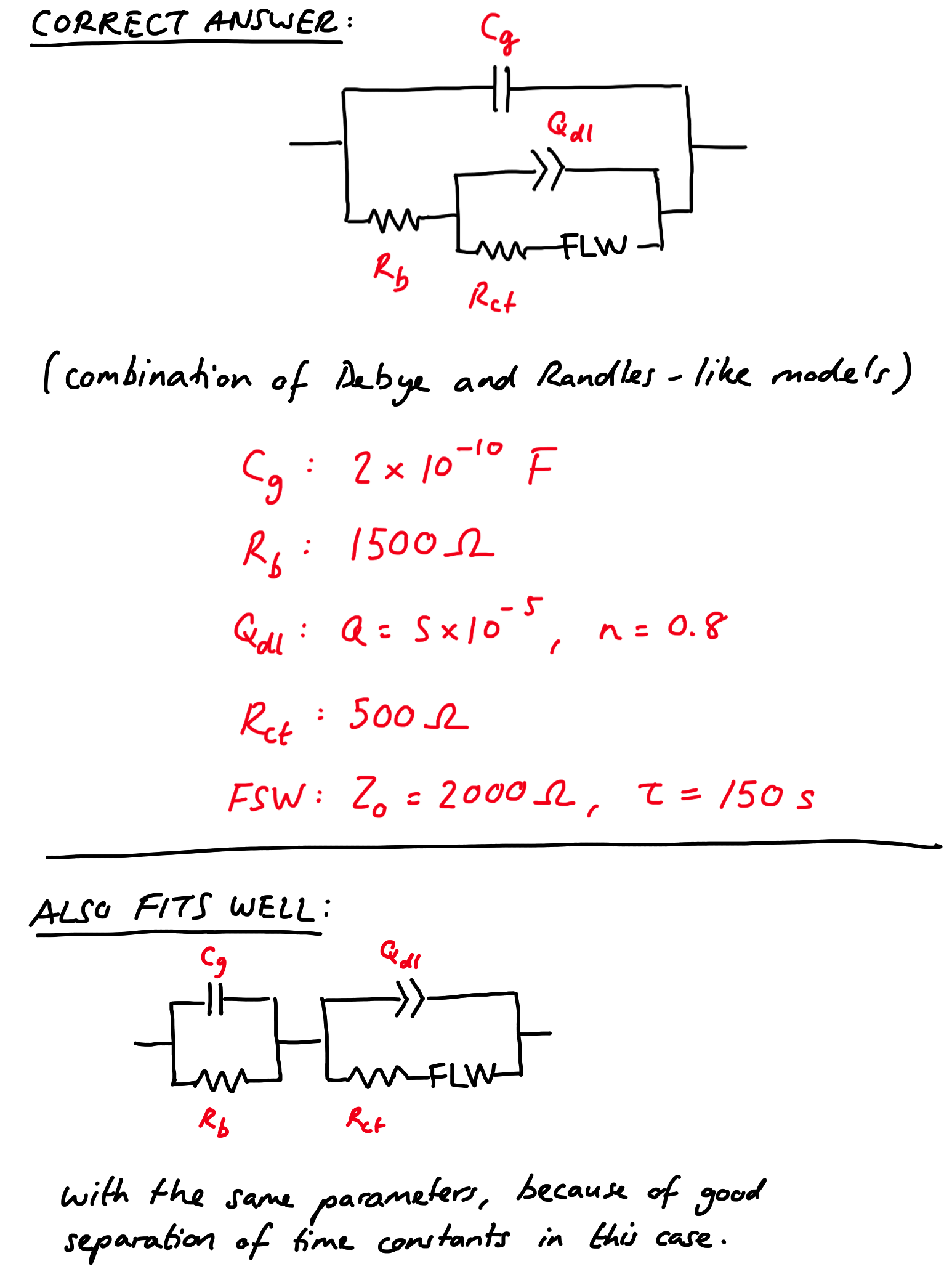{kind=link}