Chapter 1
Battery fundamentals
In Battery Fundamentals we will introduce the chemistry and electrochemistry of batteries, first in a general sense, and with a specific focus on lithium-ion (Li-ion) batteries.
In Battery Fundamentals we will introduce the chemistry and electrochemistry of batteries, first in a general sense, and with a specific focus on lithium-ion (Li-ion) batteries.
In Introduction to Battery Chemistry we will look at the fundamentals operating principles of batteries in a general sense, introducing some of the core concepts in electrochemistry.
On this page we’ll look at the origin of cell voltage using a classic battery system, the Daniell cell, as an example. We’ll look at the concepts of redox couples, standard electrode potentials, and the Nernst equation, and predict the discharge voltage curve for the Daniell cell.
Let’s start with a very simple example of a battery: the Daniell cell. This battery uses a negative electrode of zinc metal, immersed in a solution of a zinc salt, and a positive electrode of copper metal, immersed in a solution of a copper salt. Between the electrodes is a porous separator, which also separates the two salt solutions, but allows the transfer of ions between the two electrodes. A schematic of the Daniell cell is shown below.

If there is no external electronic connection between the electrodes, there is an equilibrium between the metal and the ions in the electrolyte, which we can write as a half reaction, in which the metal is the reduced form, and the dissolved ion of that metal is the oxidised form. Any pair of corresponding reduced and oxidised species is referred to as a redox couple. For each redox couple, there is an associated electrode potential, which describes the relative power of the reduced form to act as a reducing agent; that is, its tendency to release its electrons to become its oxidised form. For the Daniell cell, we have the following half-reactions:
At the negative electrode: $\ce{Zn^{2+} <=> Zn + 2e-}$, with a standard potential of -0.34 V vs SHE.
At the positive electrode: $\ce{Cu^{2+} <=> Cu + 2e-}$, with a standard potential of +0.76 V vs SHE.
The word “standard” is very important here. “Standard” means “measured under standard conditions”, which is defined as 1 mol dm-3 of dissolved species in aqueous solution, 1 atm of pressure, 25 °C. SHE means “standard hydrogen electrode”, which is the universally accepted “zero” against which standard potentials are measured. In practice, we’re usually not dealing with standard conditions, or standard electrodes, and we will deal with the consequences of this later.
The lower standard potential of the Zn/Zn2+ redox couple compared to the Cu/Cu2+ redox couple tells us that Zn metal is a stronger reducing agent than Cu; in turn, this tells us that Zn will spontaneously reduce Cu2+ ions to Cu metal, releasing energy in the process. We can write the chemical reaction as:
$$\ce{Zn + Cu^{2+} -> Zn^{2+} + Cu} \tag{1}$$Watch:
If we combine the two half-reactions shown earlier, we’ll see that the overall electrochemical reaction is the same:
$$\ce{Zn + Cu^{2+} <=> Zn^{2+} + Cu} \tag{2}$$The overall standard potential for this reaction is the difference between the positive and negative standard electrode potentials, which for this case is 1.1 V.
This tells us that if we connect the terminals of the Daniell cell to an external circuit and discharge the battery, Zn will oxidise to form Zn2+ at the negative electrode, Cu2+ will reduce to form Cu at the positive electrode, and electrons will flow from negative to positive through the external circuit. If the Daniell cell contains Cu2+ and Zn2+ solutions with concentrations of 1 mol dm-3, and is at 25 °C and 1 atm pressure, then the voltage between the terminals will be 1.1 V, minus any energy losses (which we’ll get to later).
The Daniell cell is therefore operating as a galvanic cell, which is converting the stored energy of a spontaneous chemical reaction into electricity. However, we can also force current into the cell in the opposite direction; in this case, it would operate as an electrolytic cell, where energy is required to drive a non-spontaneous chemical reaction. This process would then recharge the battery, where its stored energy can be released in the galvanic reaction at a later time.
Once we start passing current through the Daniell cell, we start moving away from standard conditions. One of the most fundamental equations in electrochemistry for calculating electrode potentials is the Nernst equation:
$$E = E^\plimsoll - \frac{RT}{nF}\ln Q \tag{3}$$In this equation, the electrode potential $E$ depends on the standard electrode potential $E^\plimsoll$ and the reaction quotient, $Q$. The reaction quotient is the product of the activities of the reaction products divided by the product of the activities of the reactants. We can write the Nernst equation for the complete Daniell cell like so:
$$E = E^\plimsoll - \frac{RT}{nF}\ln \frac{a_{\ce{Cu}}a_{\ce{Zn^{2+}}}}{a_{\ce{Zn}}a_{\ce{Cu^{2+}}}} \tag{4}$$By definition, the activities of the pure Zn and Cu metals are 1, and we can approximate the ratio of the activities of the Zn2+ and Cu2+ species by the ratio of their concentrations. We can therefore re-write the equation as:
$$E = E^\plimsoll - \frac{RT}{nF}\ln \frac{\left[{\ce{Zn^{2+}}}\right]}{\left[{\ce{Cu^{2+}}}\right]} \tag{5}$$If we plot the cell potential vs the concentration of Cu2+, we will see that the potential gradually decreases until we have almost completely depleted Cu2+ in the system:
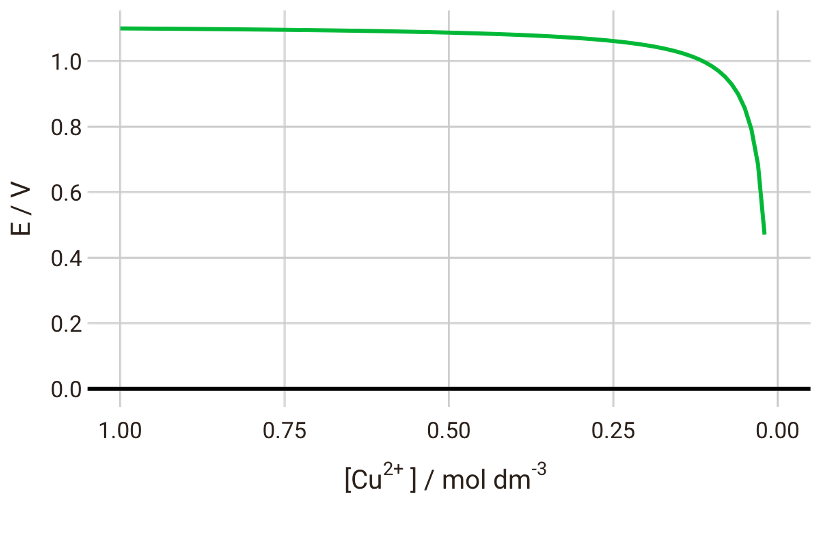
With the Nernst equation, therefore, we can predict the discharge voltage curve for this battery system.
Tables of standard electrode potentials for a huge variety of redox couples can readily be found on many websites (e.g., here) or in textbooks. In theory, any combination of two redox couples may form the basis for a battery. For example, we can find from the table of standard potentials that lithium (Li) has an extremely low standard potential of -3.01 V vs SHE, which tells us it is an extremely potent reducing agent (i.e., it really wants to be Li+). Similarly, we can find that fluorine (F) has an extremely high standard potential of 2.88 V vs SHE, which indicates that the oxidised form, F2 gas, is a very potent oxidising agent (and really wants to be F-). In principle, this would tell us that a Li–F2 battery has a theoretical cell voltage in the neighbourhood of 5.89 V, much higher than the Daniell cell. However, these numbers on their own don’t tell us much else about the properties of this combination as a battery - its rate (power) capability, rechargeability, safety, and so on. We will learn more about these factors as we continue through this guide.
In Introduction to Li-ion Battery Chemistry we will introduce the fundamental operating principles of Li-ion batteries, from thermodynamics and kinetics, to material selection, and influence of physical phenomena on device performance.
On this page we’ll take a look at the factors which determine electrode potential in intercalation electrode materials. We’ll look at the chemical potentials of the ion and the electron, and the lattice gas model: a basic model which helps us understand the origin of the different potential/voltage profile “shapes” we see in practice.
In our introduction to the thermodynamics of batteries we looked at the concept of the Nernst equation to understand what determines the electrode potential and ultimately the cell voltage. Here, we will look in more detail at the more complex chemistry which is the foundation of most Li-ion batteries, introduce a similarly fundamental model for understanding their electrode potential, and see how this helps us understand certain behaviours of these materials which we see in practice.
First, let’s remind ourselves of the basic construction of a Li-ion battery and the materials used. A schematic of a basic Li-ion cell is shown below. The arrows indicate the movement of ions and electrons when the cell is discharging.

A Li-ion battery is constructed from two electrodes, each made of a material capable of reversibly hosting and releasing Li+ ions; between them is a separator, which holds a (typically) liquid electrolyte which transfers those ions between the electrodes. The battery’s ability to store electricity relies on a large difference in the energy associated with hosting the Li+ ions in each electrode. That is, the removal of Li+ ions from the negative electrode and their insertion into the positive electrode should be accompanied by a large release of energy.
Here, we will introduce the concept of chemical potential. The chemical potential is defined as the thermodynamic ability of a particle (e.g. an atom or ion) to perform physical work. It can be thought of as the energy required to introduce one more particle to the system, or alternatively the energy obtained by removing one particle from the system.
In an intercalation material, where the Li+ ion is incorporated into the structure of a host material, the electrode potential $E$ is determined by the chemical potential of the lithium atoms in the structure $\mu_\text{Li}$, which is in turn dependent on the chemical potential of the lithium ions and the chemical potential of the electrons in the structure. Mathematically, this is written as follows:
$$E = \frac{\mu_\text{Li}}{F} = \frac{\mu_{\text{Li}^+} + \mu_{\text{e}^-}}{F} \tag{1}$$The units of chemical potential are typically kJ mol-1 and the units of Faraday’s constant are C mol-1. Dividing a chemical potential by Faraday’s constant, and accounting for a conversion of kJ to J, gives units for the potential of J C-1. One volt, 1 V = 1 J C-1.
The chemical potential of the electron is equivalent to the Fermi level, and it is always negative. Broadly speaking, the chemical potential of the electron is largely determined by the redox-active constituents in the host material: the Fermi level is defined as the energy level at which we have a 50% probability of finding an electron. In transition metal oxides, for example, this is largely determined by the redox couple of the transition metal itself, similar to what we saw in the discussion of the Daniell cell. The atoms neighbouring the transition metal in the structure also play a pivotal role.

Figure reproduced based on Goodenough and Kim, 2010
The schematic above illustrates the position of the Fermi level for some different materials. This illustrates the larger difference of the energy level of the Co3+/Co4+ couple vs Li/Li+ compared to Ti3+/Ti4+. It also illustrates the strong “inductive effect” of the PO43- anion in LiCoPO4 despite the lower oxidation states of the Co – but we will delve into this in more detail in a later page.
A simple model for the chemical potential of the ion is the lattice gas model. This model assumes that when ions are introduced into the structure they are distributed randomly with no short-range order. We can call the resulting material a solid solution - the ions ‘dissolve’ completely into the host structure without significantly altering its structure.

In the lattice gas, the chemical potential of the ion changes with the fraction of occupied sites, $x$, according to the following expression:
$$ \mu_{\text{Li}^+} = \mu_{\text{Li}^+}^\plimsoll + RT \ln \left[ \frac{x}{1-x} \right] + kx$$This right hand side of this equation consists of three parts. $\mu_{\text{Li}^+}^\plimsoll$ is the standard chemical potential for the Li+ ion in the structure. This can be thought of as the “site energy”, which is primarily determined by electrostatic interactions with the neighbouring ions in the structure. The term $RT \ln \left[ \frac{x}{1-x} \right]$ describes the contribution of the entropy of the ions in the solid solution, which is dependent on the fraction of the available sites in the structure which are occupied. Finally, we have a parameter, $k$, for modelling non-ideal interactions between the ions in the structure. $k$ is negative for attractive interactions.
We can plot this expression as a function of $x$ and animate to show the effect of changing the non-ideal parameter $k$. In the plot below, we have a y-axis of $-\frac{\left(\mu_{\text{Li}^+}-\mu^\plimsoll_{\text{Li}^+}\right)}{F}$, or the change in the chemical potential of the ion relative to the site energy, divided by Faraday’s constant. The change in this value with ion concentration in the solid reflects the change we expect in the electrode potential.
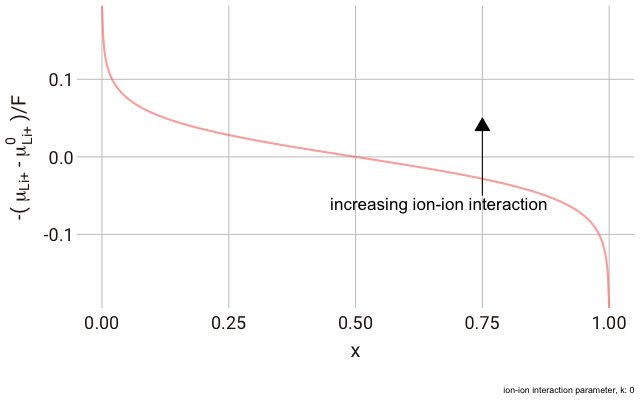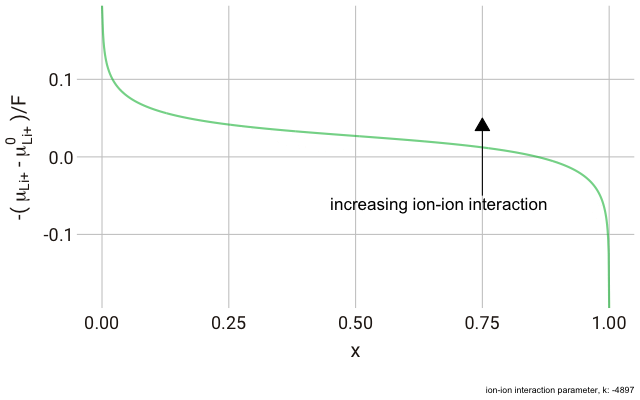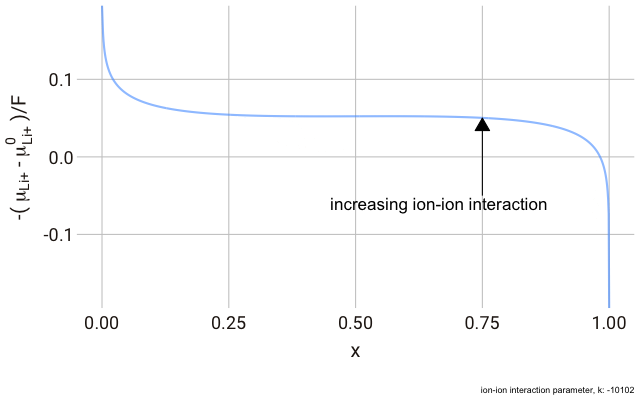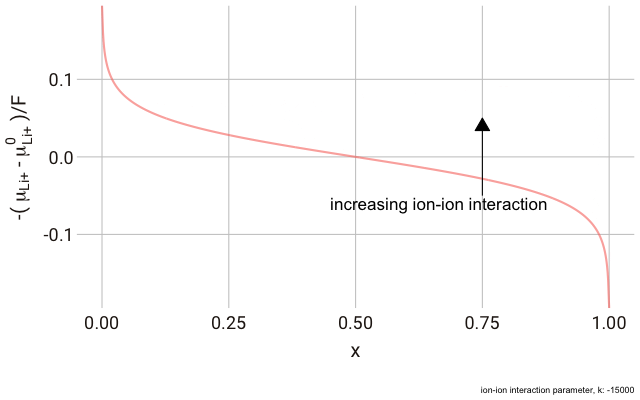
As you can see in the plot, the change in the y-axis indicates that we expect a continuously decreasing electrode potential with increasing ion content in the electrode, with the steepest change closest to the extremes of fully empty or fully occupied sites.
The decrease in $k$ (increase in ion-ion attraction) increases the slope of the curve. With a large enough $k$, the slope increases such that the value of $-\mu_{\text{Li}^+}$ begins increasing with increasing ion content, with a distinct minimum and maximum near $x = 0$ and $x = 1$. This is significant, because it predicts that with a strong enough ion-ion interaction, the material should phase-separate into two materials with high and low guest ion concentrations respectively, where the formation of distinct phases is more favourable than the solid solution. In this case, we have a “two phase” intercalation reaction.
In commercially available Li-ion battery materials, no, not really – the materials we commonly use in batteries today behave significantly differently than the ideal behaviour, but the model is still useful for understanding some of the features of intercalation materials.
That said, there is at least one class of materials which does fit the model behaviour relatively well. Chevrel phases, which are a broad class of compounds with the general formula MxMo6X8, where X is either S or Se. These compounds form slightly distorted cubic structures with very large vacancies where the guest ion M can be incorporated and be able to diffuse in all three crystallographic directions, and freely due to the ‘softness’ of the anion. The vacancies in Chevrel phases are so large that they can readily incorporate relatively large ions such as Pb2+ or Ag+. The structure of one such compound, AgMo6S8, is shown below (data for the model can be found here).
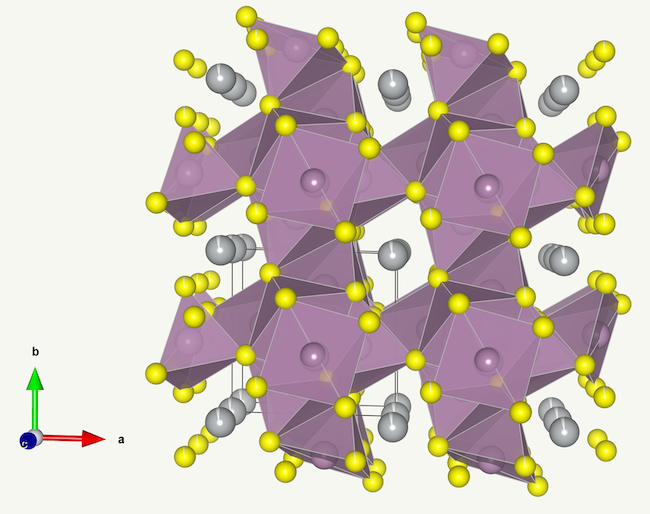
A lithium Chevrel phase, LixMo6Se8 was studied in detail in the mid-1980s([1],[2]) and demonstrated to fit the lattice gas model very closely for the composition with a Li content of up to 1 Li per Mo6Se8 unit. The fit of the model also correctly predicted the phase separation of the material into two phases at lower temperature.
[1] S.T. Coleman, W.R. McKinnon, J.R. Dahn, “Lithium intercalation in LixMo6Se8: A model mean-field lattice gas”, Phys. Rev. B 29, 4147 (1984)
[2] J.R. Dahn, W.R. McKinnon, J.J. Murray, R.R. Haering, R.S. McMillan, A.H. Rivers-Bowerman, “Entropy of the intercalation compound LixMo6Se8 from calorimetry of electrochemical cells”, Phys. Rev. B 32, 3316 (1985)
This content has been transferred from lacey.se and is not updated for this site yet.
The performance of a battery depends heavily on the properties of its electrolyte. Total ionic conductivity is one part of this, but how the current is carried by the electrolyte has some important implications, especially when the battery is subjected to very high charge or discharge currents.
Let’s consider the discharge of a Li-ion battery, containing an electrolyte with a simple salt such as LiPF6, and which is completely dissociated in the solvent. Current is drawn from the cell; Li+ ions are extracted from the negative electrode and inserted into the positive electrode. Part of this current is carried by the transport of Li+ ions, and the rest is carried by the transport of PF6- in the opposite direction:
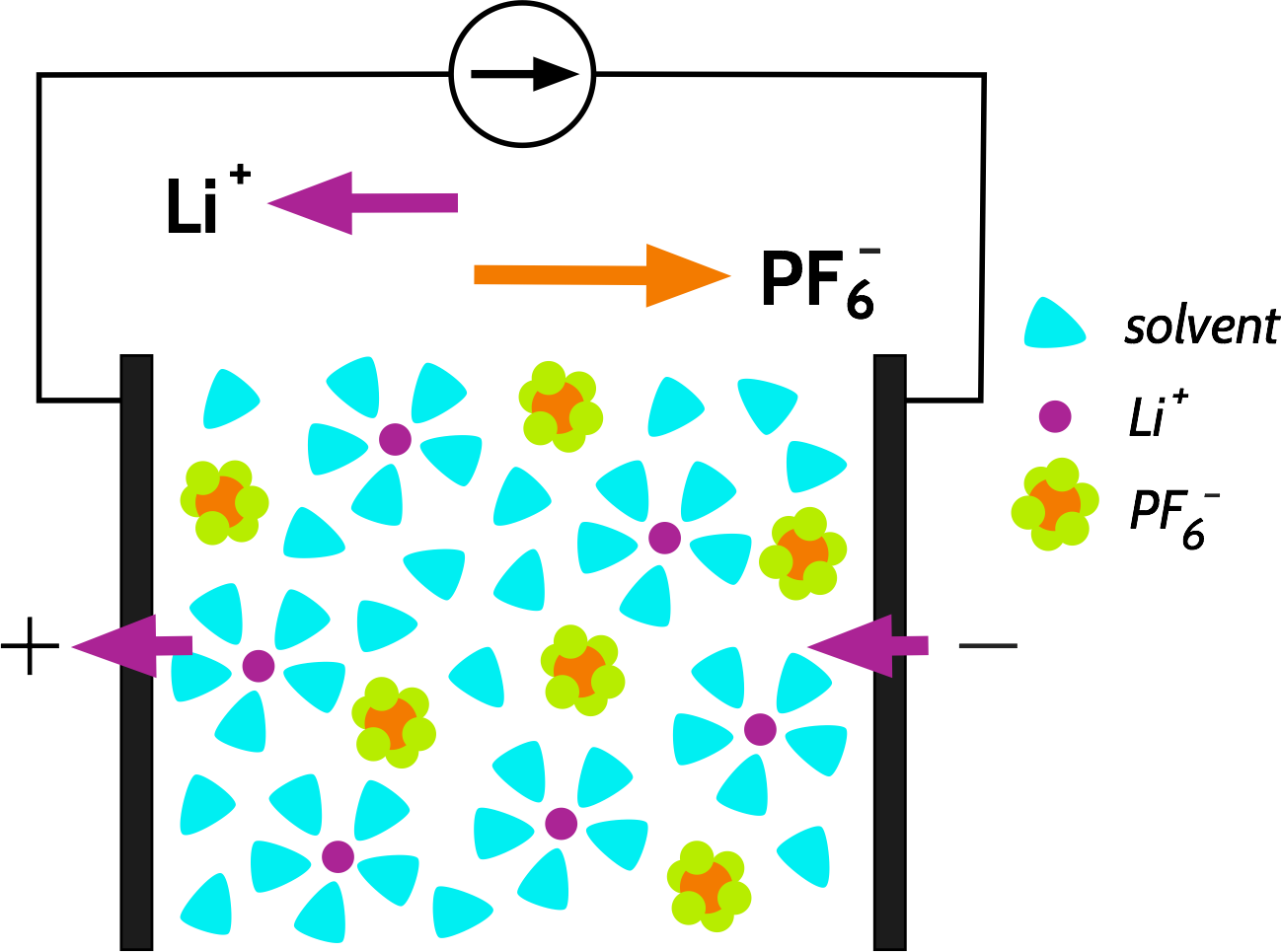
We call the fraction of the current carried by a specific species, e.g., Li+ or PF6-, the transport number, with the symbol t+ or t_ respectively. The sum of the transport numbers is 1, and by this definition, any transport number for a specific species must be between 0 and 1 (we will revisit this later). A typical value for t+ in a Li-ion battery is usually calculated to be around 0.3.
What does this mean? The transport number is effectively a ratio of the mobilities of the ions, which, following the Nernst-Einstein equation (more on this later) also relates the partial conductivities and the diffusion coefficients to each other:
$$t_+ = \frac{\mu_+}{\mu_+ + \mu_-} = \frac{\sigma_+}{\sigma_+ + \sigma_-} = \frac{D_+}{D_+ + D_-}$$t+ is often smaller than t-, because the non-coordinating anion is often more free to move in the electrolyte, while the “hard” Li+ usually has to drag a large solvation shell with it, or is relatively tightly bound to a polymer electrolyte backbone.
Back to the battery. Because only a part of the current goes into moving Li+ ions, Li+ is consumed at the interface with the positive electrode (or produced at the negative) faster than it is replenished by electrical migration. This creates a gradient in salt concentration in the electrolyte, between the electrodes of the battery. The gradient in concentration then drives diffusion of the salt, which makes up for the rest of the transport of Li+ which isn’t supplied by migration.
The formation of this concentration gradient can ultimately limit the discharge (or charge) of the battery. If the concentration of salt at an electrode surface reaches zero, the ionic resistance becomes huge, and the battery stops. If the concentration of salt becomes too high, the salt can precipitate out altogether, and the resistance can again become huge. (These processes are, however, reversible, in time.)
The value of t+, and the salt diffusion coefficient, determine how fast this concentration gradient forms, and in turn the maximum current that the electrolyte can sustain indefinitely (assuming nothing else is limiting). Both of these parameters are therefore important properties of any electrolyte, in addition to its total ionic conductivity. Ideally, t+ = 1 (and, correspondingly, t- = 0 - implying total immobilisation of the anion). In this situation, a concentration gradient cannot form at all.
Well, one important consideration, regardless of how carefully the experiment is set up, is that assumption of adherence to the Nernst-Einstein equation. This equation assumes that the ions do not interact with each other when they are dissolved, but this is only even approximately true in very dilute solution, for example concentrations < 0.01 M.
At typical salt concentrations used in batteries - 1 M, or maybe even higher - there are significant interactions between ions. Take a generic salt, LiX, for example. When we dissolve this into a solvent at a relatively high concentration, this dissociates into solvated Li+ and X- ions, but some will remain as neutral [LiX] ion pairs.
We can also consider the potential formation of ion triplets such as [Li2X]+ and [LiX2]-. Whether or not such species really exist is besides the point, but in considering these examples it is clear that the migration of the [Li2X]+ with only 1 positive charge moves two Li+; and the migration of [LiX2]- moves a Li+ in the opposite direction! These each have their own transport numbers. In this case, we should consider the concept of transference, distinct from transport. This is, in fact, the more important concept when it comes to limitations in the battery itself.
The transference number for lithium, for example, is defined as the number of moles of lithium transferred by migration per Faraday of charge. To avoid confusion with the transport number, we will use the symbol T+ (and the transference number of the anion, correspondingly, is T-). It is important to know that transference and transport are often used interchangeably, and usually with the lower case symbol t. However, they are not the same. From the above examples, it should be clear that:
$$T_+ = t_{\text{Li}^+} + 2t_{[\text{Li}_2\text{X}]^+} - t_{[\text{LiX}_2]^-}$$T+ therefore quantifies the net transference of all the Li+-containing species which migrate in the electrolyte. Of course, in an ideal system, where there is no ion association, then T+ = t+.
As with the transport numbers:
$$T_+ + T_- = 1$$However, there are no bounds on individual transference numbers. From the equation above, it is theoretically possible to have T+ < 0 if the mobility of the [LiX2]- triplet is such that it transfers Li+ in the “wrong” direction faster than the other species transfer Li+ in the “right” direction.
That’s not all though: while this is all happening, neutral [LiX] ion pairs - which do not migrate, because they are neutral - are diffusing down the concentration gradient and also, in effect, transferring Li+. However, this is not included in the definition of T+, because the definition of T+ considers only migrating species.
Ultimately, a system could have T+ < 0 and still operate, but diffusion of neutral ion pairs would have to be fast enough to both supply the current passed and compensate for the net effect of migration to transfer Li+ away from where it should be.